With the discovery of the causative gene, the disorder stands revealed as America's single most common mendelian disease. Unlike other genetic diseases, it is already curable. Indeed, genetic screening makes it potentially preventable. Yet a finding of disease-related genotype can also lead to stigmatization. Hemochromatosis therefore presents the issues surrounding genetic testing in especially stark form.
Гемохроматоз затрагивает 0,3 - 0,8% популяции американцев, т.е.
приблизительно одного человека из
каждых 200, или около 1,5 миллиона
человек. В Америке гемотохроматоз -
единственное наиболее общее
менделирующее заболевание, гораздо
более общее, чем кистозный фиброз (муковисцидоз),
серповидно-клеточная анемия или
фенилкетонурия. Врачи с большим опытом практической работы
должны встретиться с десятками или даже
сотнями случаев, которые вероятно
прошли нераспознанными или были
отнесены к другим болезням. Даже среди
пациентов с циррозом печени, ожидающим
трансплантации, у которых в трети
случаев цирроз должен был возникнуть в результате
гемохроматоза, он не прослеживается в
качестве причины заболевания. Во многих отношениях
заболевание удовлетворяет требованиям
генетического скрининга. Открытие в 1996 г.
гена хроматоза поместило это
нарушение среди сотен других, вызванных
дефектом гена или его продукта. Подобно
многим другим болезням гемохроматоз
может иметь летальный исход, но вряд ли во всех
случаях. В настоящее время заболевание
поддается лечению. При обнаружении
склонности к заболеванию генетическое
тестирование способствует предотвращению этого заболевания.
Своевременное назначение программы
периодической флеботомии или кровопускания
способствует предупреждению развития
осложнений наследственного гемохроматоза и восстановлению
нормальной продолжительности жизни.
Таким образом, течение гемохроматоза
во многом зависит от генетического
тестирования, особенно в тяжелых формах.
Но даже до генетического тестирования
существует вполне пригодный метод
обследования населения путем
чувствительного фенотипического
скрининга: определения насыщения
трансферрина (количество
сывороточного железа, деленное на величину общей
железо-связывающей способности). В
настоящее время Коллегией Американских
Патологов рекомендовано использовать
этот тест для скринирования всего
взрослого населения на наличие перегрузки
железом. Другой подход - подвергать
подобному скринированию лиц с
гетерогенными и неспецифическими
проблемами, которые потенциально могут
быть и при перегрузке железом,
включая слабость, утомляемость,
артропатии, болезни печени, болезни
сердца, или диабет. Пока ни одна из организаций не готова защищать универсальное генетическое скринирование, по крайней мере в
качестве первого теста.
Selected Reading
Bacon BR et al: Molecular medicine and hemochromatosis: At the
crossroads. Gastroenterology 116:193, 1999
Burke W et al: Consensus Statement: Hereditary hemochromatosis. Gene
discovery and its implications for population-based screening. JAMA
280:172, 1998
Cogswell ME et al: Iron overload, public health, and genetics:
Evaluating the evidence for hemochromatosis screening. Ann Intern Med
129:971, 1998
Feder JN et al: A novel MHC class I-like gene is mutated in patients
with hereditary haemochromatosis. Nat Genet 13:399, 1996
Press RD et al: Hepatic iron overload: Direct HFE (HLA-H) mutation
analysis vs quantitative iron assays for the diagnosis of hereditary
hemochromatosis. Am J Clin Pathol 109:577, 1998
Shaheen NJ, Bacon BR, Grimm IS: Clinical characteristics of hereditary
hemochromatosis patients who lack the C282Y mutation. Hepatology 28:526,
1998
Witte DL et al: Practice guideline development task force of the
College of American Pathologists: Hereditary hemochromatosis. Clin Chim
Acta 245:139, 1996
|
Gene and Protein
В пищеварительном тракте человека
железо поглощается главным образом в
дуоденальном просвете кишки. Общее поглощение
составляет 1-2 мг в день, что
уравновешивается примерно такой же
дневной потерей, в основном при
сшелушивании кожного эпителия и в
испражнениях. При гемохроматозе
поглощение железа в пищеварительном
тракте достигает 3-4 мг в день. В течение
года избыточное количество железа,
откладывающегося в клетках печени,
поджелудочной железы, pituitary, synovia и
сердца, приближается к 1 грамму. В
конечном итоге, внутри- и внеклеточный
пулы становятся насыщенными железом,
позволяя свободному железу вступать в
токсические внутриклеточные реакции.
Являясь сильным окислительно-восстановительным
веществом, железо создает свободные
гидроксильные радикалы, которые, в свою
очередь, разрушают макромолекулы
липидов, белков и даже ДНК. При средней
продолжительности жизни, прогрессирующие вследствие накопления
железа повреждения органов могут проявляться в виде дисфункции печени, диабета, hypogonadism, бесплодия, артритов,
кардиомиопатии, аритмии, гиперпигментации кожи, или
неспецифических жалоб на слабость и
утомляемость. В настоящее время
неясно что определяет смерть, цирроз,
диабет, кардиомиопатия, или гепатоцеллюлярная карцинома.
Давно известно, что генетически
гемохроматоз является менделирующим
заболеванием. Для наследования дефекта как при
рецессивном заболевании требуется наличие
сочетания двух копий генов, сцепленных с
заболеванием. Известно, что заболевание
несколько менее часто встречается у женщин, чем
у мужчин. Ген, определяющий
гемохроматоз , расположен на коротком
плече хромосомы 6, вблизи главного
локуса гистосовместимости. Так
как при наследовании признак слабо
сцеплен с HLA-A3, серологический тест на
наличие HLA-A3 часто используется для
оценки предрасположенности к
заболеванию родственников пациента с
гемохроматозом. Но так как по грубому
подсчету треть общей популяции является
положительной в отношении HLA-A3, а треть
больных гемохроматозом отрицательна,
серотипирование по HLA-A3 в значительной
степени является бесполезным для общего
скринирования на наличие гемохроматоза
или в качестве диагностического теста.
В 1996 г. ген был в конце концов найден и
выделен John N. Feder, Roger K. Wolff с коллегами
в Mercator Genetics (now Progenitor Inc.) и в Menlo Park, Calif. У огромного большинства пациентов сцепленные маркеры были
распределены широко, свидетельствуя о том, что
сцепленная с болезнью мутация возникла
в эволюции человека недавно (и,
следовательно, имела мало шансов для
изменения своего первоначального
окружения путем мейотической рекомбинации). Ограничившись
доступным отрезком на основании
совместного с маркерами наследования,
исследователи секвенировали всю
область в поисках гена-кандидата и
изучили ее на наличие мутаций.
В результате был идентифицирован ген, сцепленный с
заболеванием, нормальный продукт
которого был похож на молекулу HLA.
Первоначально ген был назван HLA-H. Когда
оказалось, что это название уже
использовано, ген переименовали в HFE.
Подобно истинному белку HLA белок HFE
простирается на клеточную мембрану. Его
внешняя часть включает нефункциональный домен, связывающий
пептид, место, где истинный белок
HLA должен захватывать антиген для
распознавания иммунными клетками. Ближе
к клеточной мембране он имеет альфа 3
петлю, место, где HFE подобно белку HLA связан с белком-соучастником, называемым бета 2- микроглобулином.
Около 80-90% больных наследственным хромотозом с
перегрузкой железом, имели в двух копиях
одного и того же гена HFE дефект:
замещение нуклеотида Г в позиции
845 на А, вызывающее замену
аминокислоты цистеина в позиции 282 на
тирозин в белке HFE(изменение, именуемое
C282Y). Потерянный цистеин должен образовывать со вторым цистеином дисульфидную связь, посредством которой
в свою очередь образовывается белковая
петля альфа 3. В мутантном белке
петля не образуется, а значит и
отсутствует способность HFE к ассоциации
с бета 2-микроглобулином.
Последний можно рассматривать как
качестве сoпровождающего или
стабилизирующего фактора. Без этого
фактора HFE не может достичь своей
нормальной локализации на мембране и
быстро разрушается. В генетических
терминах C282Y является нокаут(потеря
функции) мутацией. Вопрос,
каким образом гомолог HFE может влиять
на метаболизм железа. Механизм, посредством котoрого клетки
пищеварительного тракта захватывают
железо из просвета пищеварительного
тракта, остается неясным. Недавно
обнаружен белок DMT-1(divalent metal transporter 1), который, по-видимому, является переносчиком железа через просвет
пищеварительного тракта. Вообще метаболический путь железа (Рис.1) ведет из просвета пищеварительного тракта в intestinal-villus энтероциты, отсюда - в портальную
циркуляцию и далее в клетки мишени,
особенно в эритроидные предшественники
и гепатоциты.
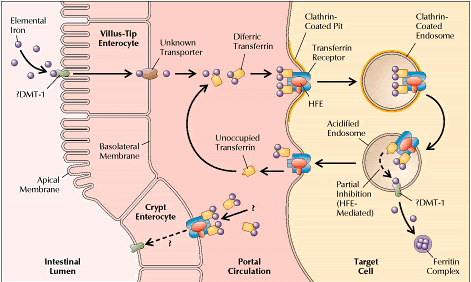 Рис.1 Детальное изображение
метаболизма железа позволяет, по
крайней мере, отчасти представить
функцию белка HFE и ее нарушение и
отсутствие при гематохроматозе. Из
просвета кишки (слева) пищевое железо
транспортируется в энтероциты вероятно
посредством недавно описанного
транспортера DMT-1. Из них железо поступает
в портальную циркуляцию чтобы затем с
помощью трансферрина быть доставленным
в клетки-мишени такие, как гепатоциты
или эритробласты (справа). Поглощенное с
помощью эндоцитоза железо в конце концов
транспортируется из эндосом в
цитоплазму часто для хранения в форме
ферритинового комплекса. Трансферрин и
его рецептор возвращаются к клеточной
поверхности. HFE связывается с транферриновым
рецептором. Связавшись, он препятствует
освобождению железа таким образом, что
увеличенная фракция связанного с
железом ферритина выходит из клетки.
В отсутствии HFE клетки становятся
перегруженными железом. Наибольшую
проблему представляет механизм
интестинального усвоения железа.
Гипотетически HFE дейтсвует в
недифференированных энтероцитах крипт (
слева внизу), в предшественниках villus-tip
энтероцитов, регулируя усвоение
плазматического железа. Каждая клетка
крипты становится чувствительной к
нагрузке организма железом, вероятно
программируя последующую экспрессию DMT-1(пунктирная
стрелка). Если функция HFE отсутствует,
чувствительность к железу исчезает. При
обманчивом ощущении низкого количества
железа в организме клетка крипты может
экспрессировать DMT-1 в избытке,
способствуя избыточному поглощению
железа из просвета зрелыми энтероцитами. Рис.1 Детальное изображение
метаболизма железа позволяет, по
крайней мере, отчасти представить
функцию белка HFE и ее нарушение и
отсутствие при гематохроматозе. Из
просвета кишки (слева) пищевое железо
транспортируется в энтероциты вероятно
посредством недавно описанного
транспортера DMT-1. Из них железо поступает
в портальную циркуляцию чтобы затем с
помощью трансферрина быть доставленным
в клетки-мишени такие, как гепатоциты
или эритробласты (справа). Поглощенное с
помощью эндоцитоза железо в конце концов
транспортируется из эндосом в
цитоплазму часто для хранения в форме
ферритинового комплекса. Трансферрин и
его рецептор возвращаются к клеточной
поверхности. HFE связывается с транферриновым
рецептором. Связавшись, он препятствует
освобождению железа таким образом, что
увеличенная фракция связанного с
железом ферритина выходит из клетки.
В отсутствии HFE клетки становятся
перегруженными железом. Наибольшую
проблему представляет механизм
интестинального усвоения железа.
Гипотетически HFE дейтсвует в
недифференированных энтероцитах крипт (
слева внизу), в предшественниках villus-tip
энтероцитов, регулируя усвоение
плазматического железа. Каждая клетка
крипты становится чувствительной к
нагрузке организма железом, вероятно
программируя последующую экспрессию DMT-1(пунктирная
стрелка). Если функция HFE отсутствует,
чувствительность к железу исчезает. При
обманчивом ощущении низкого количества
железа в организме клетка крипты может
экспрессировать DMT-1 в избытке,
способствуя избыточному поглощению
железа из просвета зрелыми энтероцитами.
На протяжении всего пути главным
внутриклеточным хранящим, и изолирующим
железо белком является ферритин, тогда
как в крови главным транпортирующим
железо белком является трансферрин. При
достижении клеток мишеней таких как
эритробласты или гепатоциты трансферрин
в комплексе с двумя молекулами железа связывается с
транферриновым рецептором, соединенным с
мембраной на поверхности большинства клеток белком. Рецептор и его лиганд в комплексе с мембраной
вдавливаются, образуя углубление
мембраны, называемое покрытой клатрином
ямкой (clathrin-coated pit), которая может
замыкаться и отшнуровываться от
мембраны. Внутри клетки получившийся
пузырек теряет свою клатриновую
оболочку и понижает свой рН, превращаясь
в закисленную эндосому. Железо освобождается от трансферрина.
Транпортированный через эндосомную
мембрану, возможно с помощью DMT-1 или
траспортера металла SFT, железо поступает
в цитоплазму для биосинтетической
утилизации (т,е. в гем) или хранится в
ферритиновом комплексе. Пузырек с
транферрином/транферрин-рецепторным
комплексом возвращается к клеточной
поверхности, где трансферрин освобождается.
В прошедшие годы стало очевидным, что белок HFE
вероятно регулирует метаболизм железа
благодаря своей способности тесно
связываться с рецептором к трансферрину ,
изменяя сродство рецептора к несущему железо
трансферрину. На основании этой
модели регуляторной функции можно предсказать,
что внутри закисленных эндосом клеток,
не относящихся к пищеварительному
тракту, в норме HFE действует, уменьшая
транспорт железа из эндосомального
HFE/трансферрин-рецепторного комплекса в
цитоплазму (Рис.2). Найдено, что у
большинства пациентов с наследственным
гемохроматозом , что помимо быстрой деградации мутантная форма HFE теряет свою способность связываться с
транферриновым рецептором. В отсутствии
этой способности транспорт железа из
эндосом в цитоплазму протекает без
отрицательной регуляции, и HFE-дефицитные
клетки (например, гепатоциты) становятся перегруженными железом.
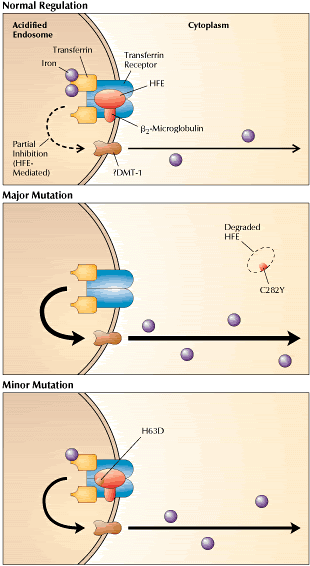 Рис.2 Главные и минорные мутации HFE обладают разным
эффектом в отношении нарушения фунуции белка HFE. Белок HFE и его стабилизирующий фактор b2-микроглобулин связываются с трансферриновым
рецептором в мембране закисленной эндосомы (как изображено в Рис.1, справа). В норме HFE отчасти подавляет освобождение железа
из трансферрина в эндосому и отсюда в клеточную цитоплазму (верхняя картинка). Главная мутация С282Y устраняет место
связывания HFE и b2-микроглобулина, способствуя деградации HFE (средняя
картинка). В отсутствии HFE транспорт железа в цитоплазму происходит без отрицательной регуляции. Минорная мутация H63D не ослабляет ни стабильность HFE,ни его связывание с трансферриновым
рецептором. Однако, мутированный белок в меньшей степени способен ингибировать эндосомальное совобождение железа(нижняя картинка). Поток железа в цитоплазму усиливается, хотя и не так сильно, как в случае наличия главной мутации. Рис.2 Главные и минорные мутации HFE обладают разным
эффектом в отношении нарушения фунуции белка HFE. Белок HFE и его стабилизирующий фактор b2-микроглобулин связываются с трансферриновым
рецептором в мембране закисленной эндосомы (как изображено в Рис.1, справа). В норме HFE отчасти подавляет освобождение железа
из трансферрина в эндосому и отсюда в клеточную цитоплазму (верхняя картинка). Главная мутация С282Y устраняет место
связывания HFE и b2-микроглобулина, способствуя деградации HFE (средняя
картинка). В отсутствии HFE транспорт железа в цитоплазму происходит без отрицательной регуляции. Минорная мутация H63D не ослабляет ни стабильность HFE,ни его связывание с трансферриновым
рецептором. Однако, мутированный белок в меньшей степени способен ингибировать эндосомальное совобождение железа(нижняя картинка). Поток железа в цитоплазму усиливается, хотя и не так сильно, как в случае наличия главной мутации.
Несмотря на то, что к числу симптомов
наследственного гемохроматоза
относятся перегруженность железом
печени, ubgjabpf, поджелудочной железы
или сердца, истинная причина -
избыточное поглощение железа - локализуеися в пределах
пищеварительного тракта. Точная роль HFE
здесь еще до конца не изучена, так как с
интестинальными клетками манипулировать трудно. Недавно возникла идея, что HFE /трансферрин-рецепторный комплекс
контролирует поглощение клетками
полученного из плазмы связанного с транферрином железа в
недифференцированных интестинальных энтероцитах, расположенных глубоко в криптах. В соответствии с этой схемой важная функция
недифференцированных клеток в криптах -
чувствовать уровень железа в крови
таким образом, что при их развитии в зрелые villus cell она может регулировать способность клеток поглощать
железо из просвета, поступающее с пищей.
В отсутствии HFE (при наследственном
гемохроматозе) железо к клеткам в криптах может доставляться
в меньшей степени, и это создает неправильное ощущение уровня
нагруженности железом. После дифференциации клетки не могут
компенсировать ошибку путем усиления
поглощения железа из просвета. В соответствии с этой пока еще не
подтвержденной гипотезой в интестинальных энтероцитах мышей с дефицитом HFE и перегрузкой железом в
печени была показана увеличенная
экспрессия предполагаемого транпортера
железа DMT-1 в просвете, который обычно
обнаруживает сверхэкспрессию только
при железо-дефицитных состояниях.
Major and Minor Mutations
После открытия HFE множество
работ было посвящено проверке первоначальной находки, касающейся
преобладания мутации C282Y у пациентов с
наследственным гемохроматозом. В
среднем от80 до 90% больных гемохроматозом
имели в своем генотипе два гена C282Y HFE и,
следовательно, были гомозиготными по
этой мутации. Даже при кистозном фиброзе,
являющемся генетически рецессивным заболеванием, наиболее
общий генетический дефект отвечает не более, чем
за 70% мутаций, сцепленный с болезнью. Высокая частота
мутации C282Y HFE при наследственном
гемохроматозе делает гентическое
тестирование более легким и экономичным,
так как проверка 845 нуклеотида в HFE
гораздо проще, чем исследование всего
гена в отношении большого разнообразия дефектов.
Среди 10% больных наследственным
гемохроматозом, не являющихся генотипически гомозиготными по
мутации C282Y, около трети, по-видимому,
имеют так называемые минорную мутацию, H63D,
в которой нуклеотид Г в 187 позиции гена HFE замещает Ц, вызывая замену аминокислоты гистидина в
63 позиции в белке HFE на аспартат. Мутация Н63D не
влияет на связь HFE с бета 2-микроглобулином
или на экспрессию HFE на клеточной
поверхности. Однако, мутация делает
белок HFE несколько менее эффективным
в регуляции взаимодействия рецепторов к транферрину с
трансферрином. Мутация H63D встречается примерно
у 25% от общей популяции , ее можно рассматривать
как "нормальную" генетическую
вариацию, или случай полиморфизма. По этой
причине положительное значение оценки
минорной мутации для идентификации гемохроматоза снижено, даже если найдено, что тестируемый субъект является гетерозиготой, несущей в
компаунде одну копию H63D и одну
копию C282Y. Установлено, что для таких лиц
перегрузка железом будет наблюдаться не
более, чем в 1% случаев.
Вообще, в популяции белых около 0,5% людей являются
гомозиготами по главной мутации HFE гена
C282Y, и 13% гетерозиготных носителей.
Превалирование главной мутации среди
некавказцев (африканцев, испанцев, ассианцев) значительно ниже, подтверждая известную этническую специфичность наследственного гемахроматоза. Около 2% общей популяции являются компаундными
гетерозиготами HFE. Эти статистические данные (Рис.3) указывают на роль генетического тестирования при любом скринировании популяции или оценке симптомов.
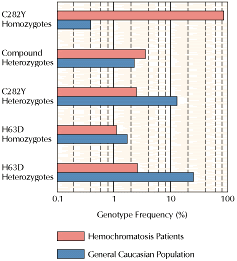 Рис.3 Распределение генотипов среди пациентов с гематохроматозом и в общей
популяции. Оно иллюстрирует контрастирующие роли главной и минорной мутаций, в возникновении заболевания.
Гомозиготными по главной мутации C282Y
являются 90% пациентов. Около трети
оставшихся , примерно 4% являются компаундными гетерозиготами, имеющими одну копию мутантного гена C282Y и одну
копию мутации H63D. Однако компаундные
гетерозиготы по частоте близки к
нормальному контролю (частоте в общей
популяции). Только у небольшого меньшинства
таких лиц(0,5-1%) будет развиваться
перегрузка железом. Подобно этому
гомозиготы H63D с приблизительно равной
частотой встречаются среди больных
гематохроматозом и в контроле. Для людей
с таким генотипом риск возникновения
перегрузки железом составляет менее 0,2%.
Гетерозиготы по одной или по другой
мутации встречаются с обычной
частотой, но имеют незначительный риск
возникновения перегрузки железом. Более,
чем 10% популяции белых являются носителями одной копии С282Y и более, чем 20% носителями одной копии H63D. Все данные
представлены графически в логарифмической шкале, и взяты из
нескольких обзоров. Рис.3 Распределение генотипов среди пациентов с гематохроматозом и в общей
популяции. Оно иллюстрирует контрастирующие роли главной и минорной мутаций, в возникновении заболевания.
Гомозиготными по главной мутации C282Y
являются 90% пациентов. Около трети
оставшихся , примерно 4% являются компаундными гетерозиготами, имеющими одну копию мутантного гена C282Y и одну
копию мутации H63D. Однако компаундные
гетерозиготы по частоте близки к
нормальному контролю (частоте в общей
популяции). Только у небольшого меньшинства
таких лиц(0,5-1%) будет развиваться
перегрузка железом. Подобно этому
гомозиготы H63D с приблизительно равной
частотой встречаются среди больных
гематохроматозом и в контроле. Для людей
с таким генотипом риск возникновения
перегрузки железом составляет менее 0,2%.
Гетерозиготы по одной или по другой
мутации встречаются с обычной
частотой, но имеют незначительный риск
возникновения перегрузки железом. Более,
чем 10% популяции белых являются носителями одной копии С282Y и более, чем 20% носителями одной копии H63D. Все данные
представлены графически в логарифмической шкале, и взяты из
нескольких обзоров.
Workup: Present and Future
Большинство экспертов склонны считать
генетическое тестирование в качестве
первичного, имеющего целью оценку риска
возникновения наследственного гемохроматоза, преждевременным. В
генетических терминах пока неизвестна
клиническая пенетрантность для
гомозогот C282Y. Для ответа
на этот вопрос требуется длительное
проспективное изучение молодых людей,
не имеющих симптомов болезни, но
гомозиготных по генотипу C282Y .
Австралийские исследователи на
основании изучения семей с гемохроматозом оценили клиническую пенетрантность гомозигот по мутации C282Y
равной примерно 80%. Практически
всегда пенетрантность у женщин ниже (
60-70%), чем у мужчин (80-90%).
При отсутствии генотипического тестирования в первую очередь врачи могут пользоваться фенотипическим
тестированием насыщения трансферрина,
рассчитываемого как величина уровня
сывороточного железа, деленная на
величину общей железосвязывающей
способности. Это тестирование также имеет
некоторую неопределенность. Уровень сыворочного
железа зависит от времени дня, диеты и
использования субъектом добавок железа.
Существуют также изменения уровня
железа,связанные с болезнью , например, из-за воспаления.
Многие из них являются истинными для
преаналитической оценки железосвязывающей способности. Однако,
в многих работах установлено, что
величина насыщения трансферрина, по
прежнему, является чувствительной и
приемлемым специфическим маркером
гемохроматоза. Наиболее определяющим
его чувствительность является выбор
верхней границы нормы. Более низкая
граница теста успешна при недостаточной
информации. При низких границах,
однако, ожидается большое число ложных
утверждений о наличии болезни. Это может
сильно увеличить объем работы по
скринированию. В 1996 г. , когда
Коллегия Американских патологов (the College of American Pathologists - CAP) рекомендовала универсальное
скринирование гемохроматоза , она установила
верхнюю границу насыщения трансферрина, равную 60% в соответствии с существующими рекомендациями
предположительно отражающую стремление
ограничить число ложных случаев. По-видимому,
выбор верхней границы будет все-таки
соответствовать несколько более низкой
величине чтобы избежать промахов. В случае
положительного теста на насыщение
транферрина алгоритм САР требует дополнительного фенотипического
тестирования перегрузки железом. Требуется, во-первых, повторное
измерение насыщения транферрина,
причем надо быть уверенным, что пациент
постился на протяжении всей предшествующей тестированию ночи. Если результат показал высокую оценку, то
в соответствии с алгоритмом рекомендуется провести следующий тест сывороточного ферритина в качестве
показателя общего железа в организме.
Высокий уровень ферритина может отражать как
перегрузку железом, так и, в некоторой
степени повреждение клеток печени.
Здесь тоже должна быть выбрана и установлена верхняя
граница нормы, но этот вопрос менее дискуссионный,
чем выбор верхней границы для уровня насыщения
трансферрином. Вероятно верхняя граница
нормального уровня ферритина должна зависеть
от пола, так как при менструации и
беременности женщины имеют механизмы
компенсации потери железа, которые не действуют у
мужчин. В сущности женщины способны к самокровепусканию -
это объясняет, почему фенотипически гемохроматоз наблюдается несколько менее часто у женщин, чем у
мужчин , хотя их генотипические частоты
идентичны. Можно также объяснить, почему
мутация C282Y в локусе HFE так сильно
распространена несмотря на то, что
фактически существует недавно. В
эволюции человека высокая
способность поглощать железо выгодна
для индивидуальной жизни в молодости,
особенно для женщин. То, что позже может
вызвать болезнь и смерть, оказывается
несущественным,
по крайней мере, для эволюции. У женщин
верхняя граница нормы для уровня
сывороточного железа вероятно
составляет 200 нг/мл, у мужчин 300-400нг/мл. Если
при повторных анализах уровни насыщения
трансферрина и сывороточного
ферритина остаются высокими, а пациент
не проявляет характерного синдрома (т.е.
воспаления, гепатита или рака), не
существует дополнительного источника,
объясняющего перегрузку железом, такого, как
переливание крови или поглощение
железа с пищей, в соответстви с
алгоритмом САР рекомендуется биопсия
печени. До 1996 г. гистопатологичекий
анализ в самом деле был существенным
основанием для определения диагноза
гемохроматоза. Алгоритм САР был
сформулирован до открытия гена HFE. После
этого возросла роль генетического
тестирования в качестве подтверждения
заболевания (Рис.4). Столкнувшись с необходимостью исследовать
фенотипические проявления перегрузки
железом, некоторые лаборатории теперь
сразу же тестируют наличие обеих
мутаций (C282Y и H63D) в гене HFE.
Наше направление - первоначальное
тестирование пациентов только в отношении
наличия мутации C282Y, так как известно,
что она в гомозиготном состоянии
является причиной заболевания у 90%
пациентов с генетическим
гемохроматозом. Пациенты,
гетерозиготные по мутации C282Y, затем
тестируются на наличие мутации H63D с
целью идентификации компаундных
гетерозигот (с низким риском
гемохроматоза), идентификации
гетерозигот H63D не делается.
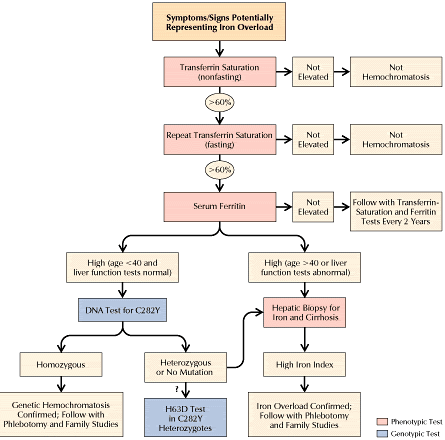
Рис.4 С введением генетического тестирования значение биопсии
уменьшается, но все же остается значительным для завершения
диагностики, сопровождающейся документальным утверждением накопления железа в органе, где оно производит свою
главную разрушительную работу. Кроме
того, биопсия имеет значение в случае
недоступности другой прогнотической
информации, а именно присутствия или
отсутствия цирроза. Если риск цирроза
низкий, биопсия теряет ценность.
Исследователи, по-видимому, близки к
заключению, что биопсия не требуется
для гомозигот по мутации C282Y, если
пациенту меньше 40 лет, ферменты печени
работают нормально и уровень сывороточного ферритина ниже 1000 нг/мл. Другими словами биопсия остается
полезной для прогноза. У пациентов,
перегруженных железом, у которых нет
мутации C282Y или есть только одна ее копия,
и у пациентов старше 40 лет с аномальными
тестами на уровни функций печени,
биопсия остается ценной и для диагноза, и для прогноза.
При использовании информации, полученной с
помощью разных тестов, главным остается
то, что осторожное решение не может быть
основано единственно на анализе
генотипа, дающем полезную информацию только в сочетании с
фенотипом. Компаундные гетерозиготы с
высоким уровнем ферритина могут быть
подвергнуты флеботомии, несмотря на
то, что сами по себе они могут и не
являться причиной гематохроматоза.
Наоборот, гомозиготы по мутации C282Y с нормальным уровнем
ферритина могут не подвергаться
флеботомии даже несмотря на высокий
риск возникновения гемохроматоза. В
самом деле пациенту настоятельно
советуют проходить периодическое
тестирование на уровень сывороточного
ферритина в крови, вероятно ежегодно или
раз в два года, и отправляться на
флеботомию, если уровень ферритина
становится ненормальным. В любом случае,
главное - это помнить о непрерывной
угрозе разрушения ткани.
О пациентах без цирроза, подвергающихся флеботомии или
кровопусканию, можно сказать, что это воздействие лечит, так
как при регулярном в соответствии с
программой применении, продолжительность жизни больный
гемахроматозом возвращается к нормальной. Даже если уже
обнаруживается фиброз или цирроз
печени, продолжительность жизни
увеличивается и ее качество
улучшается. В ответ на терапию
увеличение печени пациента становится
меньше, уменьшается с слабость и
утомляемость(хотя другие признаки
заболевания такие как артриты или гипогонадизм, как
правило не уменьшаются). Первоначально,
пациентов подвергают флеботомии один
или два раза в неделю, удаляя по 500 мл
крови, с содержанием примерно 250 мг
железа, до тех пор пока уровень
сывороточного ферритина не достигает
20-50нг/мл. Затем следует продолжать
применять флеботомию по потребности от 3 до 6 раз в год.
Dilemmas of Screening
При гематохроматозе наиболее
актуальная задача ознакомить врачей с
тем, что заболевание высоко
превалирует, требуя настороженности
большей, чем к классической триаде
болезней печени, диабетам и бронзовой
коже (skin bronzing). Цель -
предупреждение гематохроматоза.
Для гомозигот C282Y главной проблемой
является неполная клиническая
пенетрантность, оцениваемая примерно в
80%. Среди всех гомозигот, пока не имеющих
никаких симптомов, есть без сомнения
некоторые, у кого они никогда и не
возникнут, создавая дилемму, идентифицировать ли главным образом больных или всех, имеющих
соответствующий генотип. Риск возникает не для ошибочного применения лечения, в отсутствии фенотипических
аномалий флеботомию не
делают, а того, что кто-то, являющийся
гомозиготным по мутации гена гемохроматоза, может страдать от
своего клейма. И все таки большое преимущество генетического
тестирования имеет идентификация бессимптомных пациентов,
предоставляющая возможность для
предупреждения заболевания. В отсутствии фенотипических симптомов
заболевания пациентам, гомозиготным по
мутации, должно быть рекомендовано
периодическое фенотипическое скринирование. Они должны также знать, что члены их семьи имеют повышенный
риск заболеваемости и должны быть тестированы или генетически, или фенотипически, или и тем, и другим способом.
Гетерозиготы поC282Y составляют гораздо большую группу: в
кавказской популяции примерно один из
восьми. Здесь риск перегрузки железом
предельно низкий, более значительный
лишь для гетерозигот, несущих минорную
мутацию H63D во втором гене HFE (риск
достигает до 0,5-1%). Всем таким гетерозиготам необходимо дать заключение, что при вступлении в брак с
носителем один из четырех детей будет гомозиготным. Каждый ребенок одного известного носителя имеет шанс в 4%
быть гомозиготным. Существуют также
некоторые более ранние доказательства
того, что статус носительства связан с
предрасположенностью к другим заболеваниям. Носители имеют тенденцию увеличения параметров сывороточного
железа, хотя редко до патологических
уровней, и повышенный уровень железа
может быть фактором риска для возникновения некоторых форм
заболеваний печени и рака. Вообще риск
заболевания гетерозигот предельно мал,
а эта генетическая информация может
приводить к наклеиванию человеку ярлыка.
И наконец, существует проблема,
касающаяся донорства крови. Если
скринирование на мутации HFE является
универсальным, оно должно выявлять В США
от 1 до 1,5 миллионов лиц , у которых
гематохроматоз вызывает или
угрожает вызвать повреждение ткани.
Каждое лицо, включенное в программу
флеботомии, становится постоянным
источником полулитра крови примерно раз
в два месяца. Для таких пациентов
донорство должно быть доступным, давая
возможность осуществлять флеботомию по
низкой цене. Не существует доказательств, что кровь от пациентов с гематохроматозом опасна. Многи пациенты,
рано идентифицированные, должны быть
охарактеризованными как аномальные. В настоящее время большинство банков крови не принимает такую кровь.
Существует страх, что даже небольшая
выгода может сделать донорство привлекательным для лиц,
заинтересованных в злоупотреблении лекарствами IV и имеющих высокий риск отклонений сексуального поведения. Так
как скринирование на наличие гена HFE
продвигается в направлении к клинической реализации, вопрос о
донорстве крови находится среди многих
возникающих проблем, которые необходимо
разрешить.
|