Barresi, R. et al. LARGE can functionally bypass α-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nature Med. 6 June 2004 (doi: 10.1038/nm1059) | PubMed
Kanagawa, M. et al. Molecular recognition by LARGE is essential for expression of functional dystroglycan. Cell 3 June 2004 (doi: 101016/ S0092867404005434) | PubMed
Ohtsuka Y. , Kanagawa M., Yu C-C., et al. Fukutin is prerequisite to ameliorate muscular dystrophic phenotype by myofiber-selective LARGE expression. Sci Rep. 2015; 5: 8316.
Muscular Dystrophy Association Website - www.mda.org.nz Далее, www.mdausa.org - the Muscular Dystrophy Association USA website. www.muscular-dystrophy.org - the UK muscular dystrophy site. Прекрасный интерент сайт, нацеленный на помощь и информацию семей средкими нарушениями - www.nzord.org.nz Muscular Dystrophy Canada - http://www.muscle.ca. С января 2015 доступны
Сахара переносящий энзим, известный как LARGE, м. помочь в востановлении мышечной функции у пациентов с мышечной дистрофией, согласно исследованиям Kevin Campbell и др., опубликованных в Cell. Они показали, что LARGE м. осуществлять свои эффекты, обеспечивая прикрепление молекул сахара к α-dystroglycan, ключевому компоненту dystrophin–glycoprotein комплекса, который образует связь между внутренней и внешней стjронами мышечных клеток.
Muscle fibers are surrounded by a membrane that separates the inside of the fiber from the material outside the fiber — the extracellular matrix.
Most of the molecular defects that cause congenital muscular dystrophies affect proteins in the extracellular matrix, such as laminin 211, integrin, collagen 6 or alpha-dystroglycan.
The proteins known as fukutin, fukutin-related protein, POMT1, POMT2, POMGnT1, LARGE and others all participate in a special “sugar coating” (glycosylation) of alpha-dystroglycan. The sugar coating is shown as the blue, branchlike structures in this illustration. The branches are known as glycans.
Various other proteins shown — such as the sarcoglycans and dystrophin — can, when flawed or missing, cause muscular dystrophies other than CMD.
Мышечные дистрофии являются группой генетических нарушений, которые характеризуются прогрессирующим ослаблением и истощением мышц. Более тяжелыеформы CMD могут включать тяжелые умственные и речевые проблемы. По крайней мере, 30 разных типов CMD распознаются сегодня. ( Список CMD ). Большая часть типов CMD связана с белками, продуцирующими или взаимодействующими в внеклеточным матриксом, окружающим мышечные волокна. На сегодня приняты 4 категории CMD: 1. Дефекты структурных белков a.Laminin-alpha2-deficient CMD (MDC1A) b. Ullrich CMD (UCMD 1, 2, 3) c. Integrin-alpha7 deficiency (Integrinalpha7) d.CMD с булёзным эпидермолизом (Plectin) произвольных мышц (скелетных, исчерченных или поперечно-полосатых мышц. 2. Дефекты гликозилирования a. Walker-Warburg syndrome (множественные гены) b. Muscle Eye Brain disease (множественные гены) c. Fukuyama CMD (Fukutin) d. CMD + secondary laminin deficiency 1 (MDC1B) e. CMD + secondary laminin deficiency 2 (fukitin related protein deficiency, MDC1C) f. CMD с умственной отсталостью и пахигирией (мутации в LARGE, MDC1D) 3.Белки эндоплазматического ретикулума и ядра a. Rigid spine syndrome ( Selenoprotein N, 1) b. Rigid spine syndrome (Selenocysteine insertion sequence-binding protein 2) c.LMNA-deficient CMD(Laminin A/C) 4.Белки митохондриальной мембраны a.CMD с аномалиями структуры митохондрий (Choline kinase beta) Laminin-alpha2-deficient CMD (MDC1A - Merosin-deficient congenital muscular dystrophy type 1A) является наиболее распространенной CMD (~40% от всех случаев).
α-Dystroglycanopathy (α-DGP) являются гетерогенной группой мышечных дистрофий, для которых известны 15 причинных генов:POMT1, POMT2, POMGnT1, fukutin, FKRP, LARGE, ISPD, GTDC2 (POMGnT2), DAG1, TMEM5, B3GALNT2, SGK196 (POMK), B3GNT1 (B4GAT1), GMPPB, DOLK, DPM1, DPM2 и DPM3. Все они характеризуются аномальным гликозилированием α-дистрогликана, рецептора клеточной поверхности для матричных и синаптических белков, таких как ламинины, аргин, перлекан, неурексин и пикахурин. Уникальное гликозилирование O-mannosyl необходимо для связываения лигандов с помощью α-DG.α-DG взаимодействует также с трансмембранным β-DG, который в свою очередь связан с внутриклеточным дистрофином. Поэтому гликозилирование α-DG необходимо для собственно связи базальной мембраны и цитоскелета.
При таких нарушениях, α-dystroglycan, который является обычно сильно гликозилированным, лишен своих сахаров и не способен соединяться со своими лигандами во внеклеточном матриксе. Разрушение этой критической связи между внутренней и внешней сторонами мышечных клеток, как полагают, делает клетки более уязвимыми к повреждениям, индуцированным стрессами и некрозу мышечных клеток.α-дистрогликанопатии (α-DGP) включают Fukuyama congenital muscular dystrophy (FCMD), muscle-eye-brain disease (MEB), Walker-Warburg syndrome (WWS) и некоторые типы вроженных мышечных дистройий(MDCs) и limb-girdle muscular dystrophies (LGMDs).
Геном, вызывающим FCMD, является fukutin. LARGE идентифицирован как ген, ответственный за MDC1D8. Fukutin и LARGE участвуют в новой phosphodiester-linked модификации, post-phosphoryl модификации O-mannnose в α-DG. LARGE является гликозилтрансферазой, катализирующей образование повторяющихся [-3Xyl-α1,3GlcAβ1-] полимера. Эксперименты показали, что взаимодействие между LARGE и α-dystroglycan является критическим для продукции функционального, гликозилированного α-dystroglycan, а , следовательно, для поддержания мышц здоровыми. В своей работе в Nature Medicine Campbell и др. использовали LARGE для изучения, м. ли восстановление активности гликозилтрансферазы при CMD предохранять от дефектов, ассоциированных с этим субнабором мышечных дистрофий.
Они начали свой анализ, используя мышей, моделирующих CMD, которые имели мутацию в гене, кодирующем LARGE. Аденовирусы, приготовленные для экспрессии гена LARGE, инъецировали в мышцы мутантных мышей в возрасте нескольких дней. Их мышечные клетки продуцировали функциональный белок LARGE и α-дистрогликан обогащался молекулами сахаров. Способность α-дистрогликана соединяться с белками во внеклеточном матриксе восстанавливалась, и под микроскопом мышцы выглядели нормальными и здоровыми. Более того, мыши, экспрессирующие перенесенный ген страдали значительно меньше от мышечных повреждений, индуцируемых упражнениями, чем контрольные. Избыточная экспрессия LARGE увеличивает также связывание α-дистрогликана с внеклеточным матриксом в мышцах нормальных, здоровых мышей, не вызывая при этом аномалий.
Затем тестировали эффекты LARGE в клетках пациентов с CMD. Они обрабатывали клетки пациентов с тремя разными типами болезни CMD - Fukuyama CMD, muscle-eye-brain disease и Walker-Warburg syndrome - аденовирусами, которые несли ген LARGE. Хотя эти болезни вызываются мутациями в разных энзимах гликозилтрансфераз, экспрессия LARGE генерировала функциональные, гликозилированные α-дистрогликаны во всех 3-х случаях.
Эти результаты указывают на то, что стимулирующее действие добавления сахаров к α-дистрогликану с помощью LARGE м. стать потенциальным лечением для определенных мышечных дистрофий, независимо от типа мутантного энзима гликозилтрансферазы. Однако, избыточная экспрессия LARGE в клетках, лишенных GTDC2 или активности POMT1, не вызывает гипергликозилирования α-DG. Кроме того, некоторые исследования не выявили эффекта избыточной экспрессии LARGE на мутантных по POMGnT1 или FKRP мышей, а некоторые выявили вредные эффекты на эти формы болезни. Исследование терапевтических эффектов избирательной для мышечных фибрилл экспрессии гена у Large- или fukutin-дефицитных мышиных моделей α-DGP показало, что fukutin является обязательным условием для зависимого от LARGE восстановления гликозилирования α-DG (Ohtsuka et al. 2015).

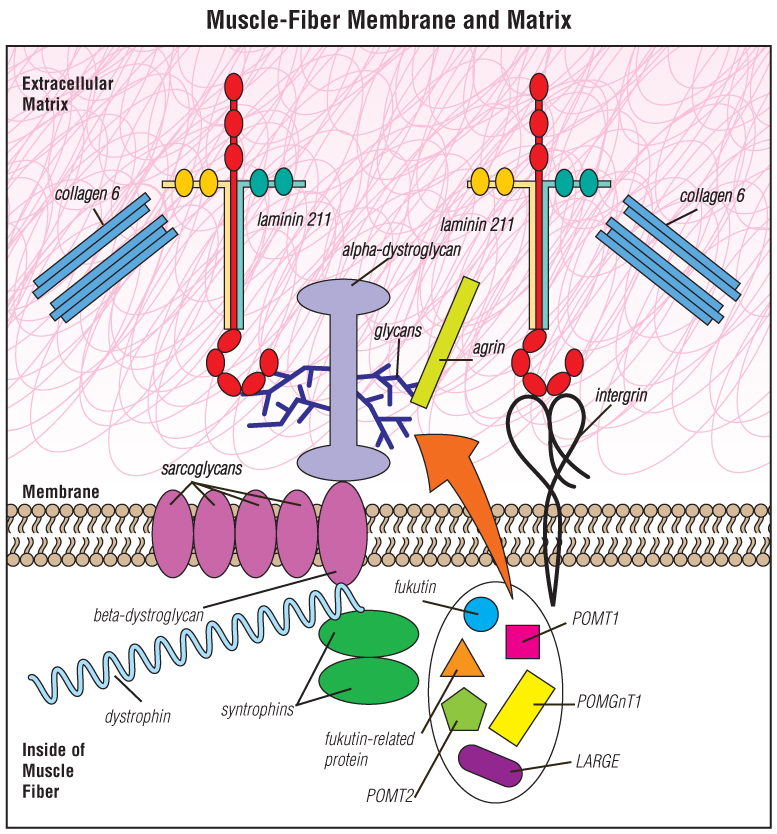 Muscle fibers are surrounded by a membrane that separates the inside of the fiber from the material outside the fiber — the extracellular matrix.
Muscle fibers are surrounded by a membrane that separates the inside of the fiber from the material outside the fiber — the extracellular matrix.