Макрофаги, обитающие в тканях центральной нервной системы (ЦНС), существуют в различных анатомических локализациях, где они выполняют различные контекстно-зависимые функции1. За пределами паренхимы макрофаги, ассоциированные с ЦНС (CAMs), обнаруживаются на поверхности головного мозга в виде лептоменингеальных макрофагов, макрофагов твердой мозговой оболочки или периваскулярных макрофагов, а также в сосудистом сплетении4-6. Считается, что CAMs контролируют целостность границ, например, во время инсульта или аутоиммунного воспаления7-9. Напротив, паренхиматозная микроглия поддерживает функцию соседних клеток, таких как олигодендроциты10 или нейроны, продуцируя трофические факторы, такие как нейротрофический фактор головного мозга11 и инсулиноподобный фактор роста 1 (IGF-1)12, а также путем быстрого уничтожения апоптотических клеток и нефункциональных синапсов13.
Нейрональные липиды представляют собой важный класс биомолекул с широким спектром важных биологических функций и высоким структурным разнообразием, определяющим их расположение в клетках. Например, в то время как жирные кислоты, триглицериды и стероловые липиды в основном локализованы в органеллах нейрональных клеток, сфинголипиды, такие как сфингомиелин и гликосфинголипиды, в основном находятся на плазматической мембране нейронов. Там ганглиозиды, как типичные гликосфинголипиды, составляют 10-12% от общего содержания липидов и образуют мембранные микродомены ("липидные плотики") с различными клеточными функциями, такими как передача сигналов, эндоцитоз и мембранный транспорт14. В процессе развития состав ганглиозидов головного мозга меняется от преимущественно простых (GM3) к сложным (GM2, GM1) ганглиозидам, что указывает на потенциальную роль ганглиозидов в развитии головного мозга15. Участие микроглии в процессе обмена нейрональных гликосфинголипидов в значительной степени неизвестно.
Недавнее транскриптомное профилирование выявило широкий спектр генов, ассоциированных с микроглией6,16,17, включая Hexb, который кодирует β-субъединицу (HEXB) димерного лизосомального фермента β-гексозаминидазы (Hex). Этот фермент катализирует гидролиз концевых остатков N-ацетил-гексозамина в различных гликоконъюгатах, включая гликолипиды. Существуют два основных изофермента Hex: гетеродимерный Hex A, состоящий из одной α- и одной β-субъединиц, и гомодимерный Hex B, состоящий из двух β-субъединиц18. Только Hex A разрушает ключевой физиологический субстрат - ганглиозид GM219. У людей наследственный дефицит HEXB вызывает болезнь Сандхоффа20. Хотя генетическая причина и накопление GM2 хорошо установлены, специфический клеточный вклад в клиренс ганглиозидов в ЦНС остается неясным. Мы предположили, что микроглиальный Hex необходим для гомеостаза ганглиозидов, и его дефицит может вызывать нарушение деградации GM2 во всей ЦНС, способствуя развитию болезни Sandhoff. Соответственно, мы интегрировали различные многомерные транскриптомные и липидомные методы для изучения оборота ганглиозидов в CNSs мыши и человека. Кроме того, мы создали мутанты, специфичные к типу клеток, и, используя различные химерные системы переноса, описали критическую ось микроглия-нейрональный Hex-GM2–макрофагальный лектин галактозного типа 2 (MGL2), которая необходима для поддержания гомеостаза ЦНС.
Hexb is a stable microglia gene
Недавние исследования были направлены на идентификацию генов микроглии, которые надежно отделяют их от CAMs или других клеток-резидентов мозга, нацеленных конкретно на эти клетки.
21,22 Ключевые гены включают
P2ry12, Tmem119, Sall1 и Hexb. Чтобы идентифицировать гены микроглии, которые активно экспрессируются при патологии, мы провели секвенирование одиночных ядер 3'-мРНК (snRNA-seq) микроглии в пяти моделях нейродегенерации или демиелинизации. Как и ожидалось, появилось несколько контекстно-зависимых кластеров микроглии (рис. 1а). Среди основных генов микроглии
Hexb, P2ry12 и Cx3cr1 демонстрировали стабильно высокую экспрессию, тогда как уровни
Tmem119, Sall1, Gpr34, Siglech, Olfml3 и Fcrl2 были либо низкими, либо вариабельными (рис. 1b).
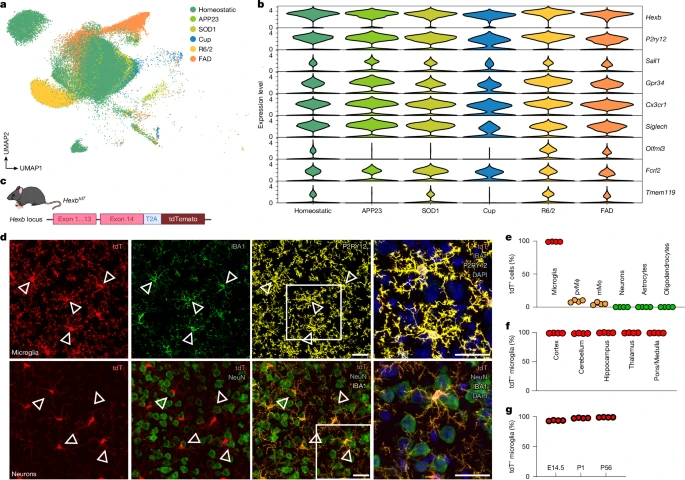 Fig. 1: Hexb expression in the mouse brain is highly microglia-enriched throughout CNS conditions, brain regions and development.
Fig. 1: Hexb expression in the mouse brain is highly microglia-enriched throughout CNS conditions, brain regions and development.
Чтобы изучить влияние Hexb на здоровье, мы воспользовались нашим репортером HexbtdT line23 (рис. 1с). Следует отметить, что практически все кортикальные микроглии экспрессировали tdTomato (99,06 ± 0,10%), в отличие от периваскулярных макрофагов (9,10 ± 0,89%), лептоменингеальных макрофагов (5,04 ± 0,99%), нейронов (0,01 ± 0,01%), астроцитов (0,03 ± 0,03%) и олигодендроцитов (0%) (рис. 1d)., e и расширенные данные Рис. 1а). Более того, микроглия была tdTomato+ во всех областях мозга и сохраняла экспрессию Hexb на протяжении всего развития (рис. 1f, g и расширенные данные рис. 1b–d). Скрещивание мышей HexbtdT с Cx3cr1GFP или Thy1GFP подтвердило отсутствие перекрытия микроглии, экспрессирующей Hexb, с GFP+ CAMs или нейронами, соответственно (расширенные данные рис. 1e,f). Предыдущие сообщения о низком содержании мРНК Hexb в нейронах мышей24 были подтверждены у взрослых мышей дикого типа (данные не приведены). В целом, Hexb является высокостабильным геном микроглии в ЦНС мыши во время развития, гомеостаза и заболевания.
Early and robust microglial activation
Доказав, что микроглия экспрессирует
Hexb на высоком уровне в мозге мыши, мы затем исследовали его функциональную роль. У мышей с нокаутом по
Hexb (
Hexb-/-), ранее созданных в качестве модели болезни Сандхоффа
25, развились быстро прогрессирующая атаксия и потеря веса при сохранении силы захвата, и они умерли на 114 ± 12 день после рождения (P) (рис. 2a,b и расширенные данные рис. 2a). Примечательно, что в крови не было обнаружено периферических воспалительных процессов, вызывающих энцефалопатию, вызывающую двигательные симптомы (расширенные данные рис. 2б, в), а в головном мозге отсутствовали лимфоцитарные инфильтраты (данные не показаны). В отличие от этого, в микроглии наблюдались заметные изменения в количестве и морфологии (рис. 2c–f и расширенные данные рис. 2d). Количество клеток микроглии увеличилось на 28% и достигло максимума на 85%, что указывает на раннее поражение до начала клинических проявлений. Плотность варьировала в зависимости от региона, при этом наибольшие показатели были зафиксированы в таламусе, мосту/продолговатом мозге и белом веществе мозжечка (рис. 2с). Трехмерная (3D) реконструкция лизосом IBA1+ микроглии и CD68+ показала увеличенный объем лизосом, меньшее количество терминальных точек/ответвлений и укороченные отростки (рис. 2d, e). Более того, микроглия повышала активность маркера активации лизосом Mac-3 уже в возрасте 7 лет (рис. 2g и расширенные данные рис. 2d–f) и снижала активность TMEM119 и P2RY12, подчеркивая их активированный фенотип. Потеря P2RY12 была наиболее заметна в таламусе (51,29 ± 4,23% IBA1+ микроглии) (расширенные данные, рис. 2g). Астроглиоз появился только на P85, когда мыши уже были поражены клинически (рис. 2h и расширенные данные рис. 2d), тогда как внеклеточные отложения APP+, признак повреждения аксонов, появились поздно, особенно в таламусе (рис. 2c, i и расширенные данные рис. 2d). Таким образом, недостаток Hexb приводит к ранней и чрезмерной активации микроглии, что проявляется выраженными численными, морфологическими и лизосомальными изменениями, в то время как астроглиоз и повреждение аксонов являются поздними проявлениями.
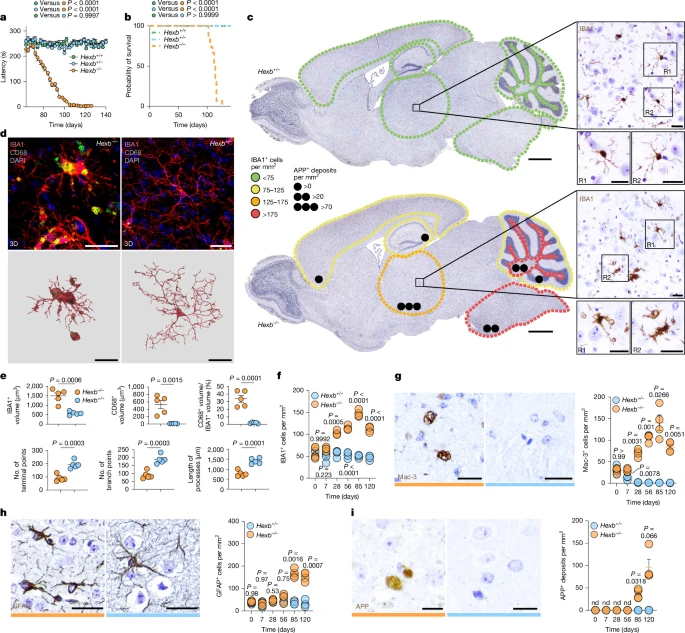 Рис. 2. Широко распространенная и выраженная лизосомальная активация микроглии на ранней стадии является ключевой особенностью патологии, опосредованной Hexb.
Рис. 2. Широко распространенная и выраженная лизосомальная активация микроглии на ранней стадии является ключевой особенностью патологии, опосредованной Hexb.
Molecular census of Hexb -/- CNS cells
Чтобы изучить молекулярные основы активации микроглии у мышей
Hexb-/-, мы провели snRNA-seq ядер таламуса на ст. P7 и P120. Контролем служили гетерозиготные мыши (Hexb+/-), поскольку у них не было различий в экспрессии по сравнению с микроглией Hexb+/+ (дополнительный рис. 1). Таламус был выбран для транскриптомного профилирования из-за выраженного микроглиоза, серьезного повреждения аксонов и его известного участия в развитии болезни Сандхоффа у человеков
26. После контроля качества мы проанализировали 103 201 ядро и определили типы клеток, используя как инструмент Azimuth, так и эталонные наборы генов9 (рис. 3а и расширенные данные Рис. 3а, б). Доля иммунных клеток заметно увеличилась у пожилых мышей
Hexb-/- (рис. 3b и расширенные данные на рис. 3c, e). В иммунной популяции мы выявили семь различных кластеров микроглии. Кластеры c0 и c1 включали практически всю взрослую контрольную микроглию, таким образом, представляя собой "гомеостатические" кластеры. Кластеры c2–4 были чрезмерно представлены в выборках с заболеваниями, в то время как в неонатальной микроглии (c5) или CAMs отсутствовали кластеры, связанные с заболеванием, что указывает на участие в заболевании исключительно микроглии (рис. 3c и расширенные данные рис. 3d, f). Анализ псевдовремени выявил траекторию от переходного кластера c2 к кластерам c3 и c4 с терминальными заболеваниями (рис. 3c). Эти кластеры показали снижение уровня гомеостатических маркеров (
P2ry12, Cx3cr1, Gpr34, Sall1 и Siglech) и увеличение количества генов активации
27 (
Apoe, Ctsb, Lyst, Csf1, Tyrobp, B2m, Spp1 или Cybb) (расширенные данные на рис. 3d). Кластеры c2 и c3 разделяли повышенную регуляцию
Igf1, Apobec1 и Flt1 (рис. 3d, e,g). Анализ генной онтологии (GO) связал c2 с терминами, связанными с цитокинами, c3 - с путями аутофагии/фагоцитоза, а c4 - с реакциями на интерферон (рис. 3d, f). По сравнению с другими моделями нейродегенерации (рис. 1а), микроглия с дефицитом
Hexb имела общие закономерности, но наиболее точно соответствовала кластерам 11 и 12 у мышей 5xFAD и APP23 (расширенные данные, рис. 3g–k). В совокупности потеря
Hexb вызывает прогрессирующую активацию микроглии, которая приводит к специфичному для заболевания, крайне дисфункциональному состоянию, характеризующемуся изменением иммунного, аутофагического и фагоцитарного профилей.
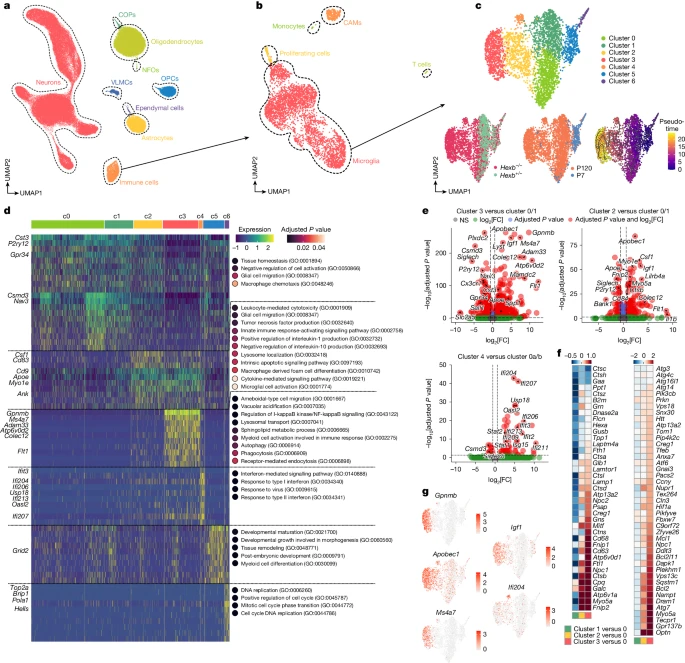 Рис. 3. Молекулярный анализ мозга мышей с дефицитом Hexb.
Рис. 3. Молекулярный анализ мозга мышей с дефицитом Hexb.
A spatiotemporal lipid map
Чтобы оценить влияние дефицита
Hexb на липидный состав ЦНС, мы провели нецелевую липидомику гомогенатов коры головного мозга мышей
Hexb-/- и Hexb+/-. Из 5825 идентифицированных липидов 196 были значительно повышены, а 59 - снижены в
Hexb-/- головном мозге (рис. 4а). Важно отметить, что количество различных видов GM2 было заметно увеличено, в то время как количество продуктов, полученных из Hex, ганглиозидов GM3, значительно сократилось (расширенные данные на рис. 4а). Чтобы составить карту пространственного распределения ганглиозидов, мы применили нецелевую масс-спектрометрию с использованием лазерной десорбции/ионизации (MALDI)
28 к срезам головного мозга в разные моменты времени. Некоторые физиологически регулируемые виды липидов изменялись с возрастом (например, кардиолипины, сульфатиды (SM4), ганглиозиды (GT1, GR3, GD1, GD3)), но не различались между генотипами (расширенные данные рис. 4с). Напротив, накопление GM2 уже было обнаружено при P0 в мозге нокаутированных пациентов (рис. 4b, d и расширенные данные рис. 4b). Примечательно, что молекулы GM2 с более короткими жирными ацильными цепями (например, d34:1) преимущественно накапливались у новорожденных, тогда как молекулы GM2 с более длинными жирными ацильными цепями (например, 38:2) были обогащены при P120 (рис. 4b,d). Этот сдвиг был особенно выражен в таламусе, где наблюдалось заметное накопление GM2 на ст. P120 (рис. 4c и расширенные данные на рис. 4d). Внутри таламуса GM2 локализован в определенных ядрах — центромедианном и парафасцикулярном, ключевых источниках таламостриатальных проекций, участвующих в управлении двигательной активностью
29 (рис. 4с). Помимо GM2, патологически повышенные уровни липидов включали GA2, asialo-GM2 и бис (моноацилглицеро) фосфат (BMP) в соотношении 44:12 (дополнительный рис. 2). Иммунофлуоресцентная визуализация подтвердила наличие GM2 в нейронах и микроглии, но не в астроцитах и олигодендроцитах (расширенные данные на рис. 4е). Как микроглия, так и нейроны демонстрировали значительную нагрузку лизосомальными ганглиозидами, причем нейроны имели тенденцию к более высоким уровням (расширенные данные на рис. 4f, g). В целом, наша пространственная карта ганглиозидов выявила отчетливый липидный профиль, связанный с заболеваниями, обусловленный потерей Hexb и региональным накоплением GM2, особенно в таламусе.
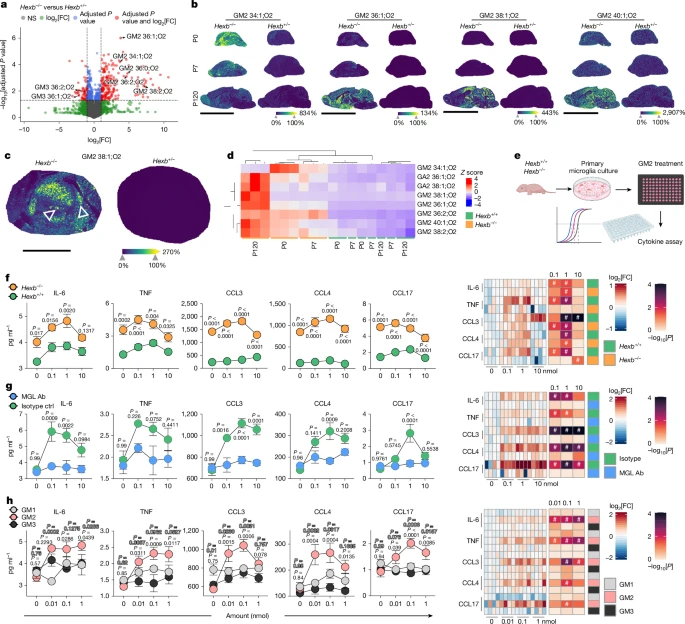 Рис. 4. Отсутствие Hexb приводит к характерному пространственно-временному накоплению GM2, которое индуцирует выработку микроглией провоспалительных цитокинов через MGL2.
Рис. 4. Отсутствие Hexb приводит к характерному пространственно-временному накоплению GM2, которое индуцирует выработку микроглией провоспалительных цитокинов через MGL2.
GM2 activates microglia by MGL2 engagement
Определив, что GM2 является наиболее нарушенным липидом в Hexb-/- головном мозге и его пространственное совпадение с микроглиозом, мы исследовали его прямое воздействие на микроглию. Первичную микроглию контрольных мышей Hexb-/- и Hexb+/+ помещали в 96-луночные планшеты, покрытые GM2, и измеряли секрецию цитокинов и хемокинов (рис. 4е). GM2 индуцировал дозозависимое высвобождение IL-6, TNF, CCL3, CCL4 и CCL17 с более высокими уровнями в Hexb-/- клетках, вероятно, вследствие предварительной активации при хроническом воздействии ганглиозидов (рис. 4f и дополнительный рис. 3). Чтобы разобраться в механизме, мы сосредоточились на MGL2, рецепторе лектина С-типа, экспрессирующемся на дендритных клетках, макрофагах и микроглии, который специфически связывается с концевым N-ацетилгалактозамином (GalNAc), присутствующим на GM2 (ссылка 30). Хотя известно, что MGL2 участвует в эндоцитозе31, мы проверили, опосредует ли он также активацию микроглии, управляемую GM2. Действительно, микроглия, предварительно обработанная антителом, блокирующим MGL2, больше не реагировала на GM2 высвобождением цитокинов, в то время как клетки, обработанные изотипом, реагировали (рис. 4g и дополнительный рис. 3). Чтобы исключить неспецифические эффекты антител, мы использовали EGTA для хелатирования внеклеточного кальция, необходимого для функционирования лектинов С-типа, или GalNAc для конкурентного ингибирования связывания с MGL2, оба из которых подавляли цитокиновые реакции, подтверждая специфичность передачи сигналов MGL2-GM2 (Расширенные данные на фиг. 5a–c). Кроме того, GM1 и GM3, в которых отсутствует доступный или присутствующий GalNAc, не индуцировали цитокинов32 (рис. 4h и дополнительный рис. 3).
Чтобы подтвердить наши результаты in vivo, мы вводили антитела, блокирующие MGL2, посредством внутрижелудочковых инъекций мышам Hexb-/- и Hexb+/- из P10 в течение 3 недель (расширенные данные, рис. 5d). Инъецированный Hexb-/- мозг показал значительное снижение уровня TNF и CCL4, наряду со снижением уровней IL-1α и CXCL9 (расширенные данные, рис. 5e,f). В микроглии, очищенной методом сортировки клеток, активируемых флуоресценцией (FACS), наблюдалось снижение мРНК Ccl5 и Cx3cl1 и тенденция к снижению экспрессии мРНК Il1b и Il18 (расширенные данные, рис. 5g). В совокупности эти результаты демонстрируют, что GM2 действует как специфический активатор микроглии посредством MGL2 как in vitro, так и in vivo.
Hexb loss impairs neuronal excitability
Определив механизм активации микроглии, вызванной GM2, мы затем исследовали, влияет ли дефицит Hexb и последующее накопление GM2 на передачу сигналов нейронами. Таким образом, мы выполнили электрические записи от пирамидных нейронов двигательного слоя коры 2/3 в острых срезах (расширенные данные на рис. 5h, i). Хотя входное сопротивление и мембранный потенциал не изменились (расширенные данные, рис. 5j), нейроны Hexb-/- генерировали значительно меньшее количество потенциалов действия во время этапов деполяризации тока, что указывает на снижение возбудимости (расширенные данные, рис. 5i,k). Кроме того, изменения полу-ширины потенциала действия (расширенные данные рис. 5л,м) и провала напряжения при гиперполяризации (расширенные данные рис. 5л, м). 5n) предполагает нарушение регуляции основных проводимостей. На сетевом уровне нейроны Hexb-/- показали сниженную частоту синаптических входов, что отражает нарушение глутаматергической связи (расширенные данные, рис. 5о). В совокупности эти данные свидетельствуют о значительном нарушении автономной функции нейронов и их цепей при дефиците Hexb и накоплении GM2.
Microglial and neuronal Hexb drive disease
До сих пор ограниченные средства нацеливания на конкретные типы клеток препятствовали выявлению типов клеток, вызывающих заболевание при болезни Сандхоффа. Хотя считается, что потеря нейрональной активности Hex приводит к накоплению GM2 и развитию патологии
33, высокая экспрессия
Hexb в микроглии (рис. 1c–g) указывает на дальнейшую роль в прогрессировании заболевания. Чтобы проанализировать клеточно-специфический вклад, мы расположили экзон 2 гена
Hexb по бокам от
loxP-сайтов и создали мышей
Hexbfl/fl (расширенные данные, рис. 6а). Они были скрещены с Nescre/+ (нацелены на нейроэктодермальные клетки
34, включая нейроны) и Cx3cr1cre/+ (нацелены на миелоидные клетки
6,35, включая микроглию). Каждая строка показывала эффективное удаление Hexb (расширенные данные на рис. 6b–d). Удивительно, но ни мыши Nescre/
+:Hexb
fl/fl, ни мыши Cx3cr1
cre/+:Hexb
fl/fl не повторяли фенотип, наблюдаемый у конститутивных мышей
Hexb-/-, и выживаемость, двигательная функция и масса тела оставались нормальными (рис. 5a,b и расширенные данные рис. 6e,f). У обоих пациентов с одиночным нокаутом общая активность Hex в мозге была лишь незначительно снижена (рис. 5с), а нейроны сохраняли ферментативную активность и HEXB-позитивные структуры, что указывает на избыточную роль экспрессии
Hexb в микроглии или нейронах (рис. 5d–f и расширенные данные рис. 6g). Примечательно, что только у мышей с двойным нокаутом
Cx3cr1cre/+:Nescre/+:Hexbfl/fl заболевание повторилось с поздним появлением двигательных симптомов, потерей веса и преждевременной смертью (рис. 5a,b и расширенные данные рис. 6e). У этих мышей наблюдалось заметное снижение активности Hex и массовая потеря нейронов HEXB+ (рис. 5c, e, f и расширенные данные на рис. 6g). Микроглия также имела аномальную морфологию и увеличенное количество клеток (рис. 5g). В целом, для развития болезни Сандхоффа достаточно только комбинированного дефицита Hexb в нейроэктодермальном и миелоидном отделах.
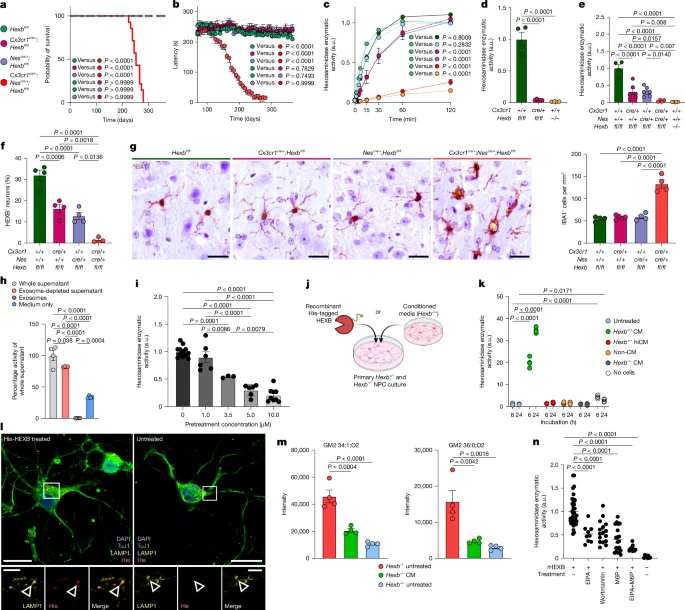 Рис. 5. Совместная секреция микроглии и поглощение Hex нейронами поддерживают гомеостаз ЦНС и предотвращают болезнь Сандхоффа.
Рис. 5. Совместная секреция микроглии и поглощение Hex нейронами поддерживают гомеостаз ЦНС и предотвращают болезнь Сандхоффа. Microglia aid neuronal lysosomal function
Поскольку ни микроглия, ни нейроны сами по себе не вызывают заболевания, мы предположили, что существует компенсаторный механизм обновления GM2, включающий высвобождение ферментов микроглии. Чтобы проверить это, мы культивировали микроглию Hexb+/+ и измерили активность Hex в супернатанте (расширенные данные на рис. 7а). Активность была значительно выше, чем в контрольной группе, получавшей только среду, и локализовалась во фракции, обедненной экзосомами, что указывает на секрецию свободного Hex (рис. 5h и расширенные данные рис. 7b). Чтобы определить путь секреции, мы обработали микроглию различными ингибиторами (рис. 5i и расширенные данные на рис. 7c–f). Среди них только golgicide A and brefeldin A значительно и дозозависимо блокировали секрецию Hex микроглией (рис. 5i и расширенные данные рис. 7d), что позволяет предположить высвобождение посредством классического секреторного пути. Thapsigargin и BAPTA-AM также снижали секрецию фермента, вовлекая внутриклеточный Ca2+ в устойчивую секрецию, тогда как vacuolin-1 и EGTA не оказывали никакого эффекта (расширенные данные на рис. 7e,f).
Для оценки усвоения нейронами Hexb-/- и Hexb+/- нейральных клеток-предшественников (NPC) обрабатывали рекомбинантным Hex, меченным His, или кондиционированной средой из первичной микроглии дикого типа (рис. 5j и расширенные данные рис. 7g). Кондиционированная среда вызывала зависящее от времени повышение ферментативной активности у NPC (рис. 5k и расширенные данные на рис. 7h), подтвержденный экспериментами по совместному культивированию Transwell, подтверждающими, что перенос фермента не зависит от прямого контакта с клетками (расширенные данные, рис. 7i). Вестерн-блоттинг подтвердил поглощение Hex NPC (расширенные данные, рис. 7j), а иммунофлуоресцентная визуализация подтвердила лизосомную локализацию интернализованного фермента (рис. 5l). Примечательно, что обработанные кондиционированной средой Hexb-/- нейроны сохраняли меньше GM2, что подтверждает функциональный вклад Hex, полученного из микроглии, в деградацию нейрональных ганглиозидов (рис. 5м и расширенные данные рис. 7к).
Чтобы изучить механизмы поглощения, мы совместно лечили Hexb-/- NPC ингибиторами: 5-(N-этил-N-изопропил) amiloride (EIPA) и Wortmannin (микропиноцитоз) или маннозо-6-фосфатом (блокада рецепторов M6P). Все они значительно снижали поглощение Hex (рис. 5n), это указывает на два параллельных пути: макропиноцитоз и эндоцитоз, опосредованный M6PR. Эти результаты были дополнительно подтверждены на Hexb-/- фибробластах (расширенные данные на рис. 5n). 7l–p).
Чтобы протестировать этот механизм в тканях, мы истощили микроглию в органотипических срезах гиппокампа Hexb-/- или Hexb+/- с помощью clodronate, затем повторно ввели микроглию, компетентную к Hexb (расширенные данные, рис. 7q). При истощении активность Hex полностью снижалась и сильно коррелировала с количеством микроглии IBA1+ на 17-й день, что явно указывало на микроглию как на основной источник секретируемого Hex в ЦНС мыши (расширенные данные на рис. 7r, s,u). На 17-й день мы наблюдали HEXB в нейронах химерных Hexb-/- срезов, что подтверждает наличие микроглии (расширенные данные, рис. 7т). В совокупности микроглия выделяет Hex по классическому секреторному пути, зависящему от Гольджи. Фермент эндоцитозируется нейронами посредством макропиноцитоза и эндоцитоза, опосредованного рецепторами M6P, переносится в лизосомы и способствует деградации GM2.
Hexb in MLCs prevents neurodegeneration
Продемонстрировав, что микроглия может восстанавливать нейрональную активность Hex
ex vivo, мы затем проверили, может ли замена микроглии обеспечить терапевтический эффект
in vivo. С этой целью мышей
Hexb-/- подвергали истощению микроглии с помощью ингибитора CSF1R BLZ945 с последующей трансплантацией костного мозга Cx3cr1
GFP/+:Hexb
+/- (фиг. 8a–d). Мыши-реципиенты показали высокую степень приживления миелоидных клеток после P245 (рис. 6a,b и расширенные данные на рис. 8e). Примечательно, что химерные мыши
Hexb-/- продемонстрировали нормальную выживаемость, смягченные двигательные симптомы и стабильную массу тела (рис. 6c–e и расширенные данные рис. 8f). Гомогенаты головного мозга демонстрировали восстановленную активность Hex и заметно сниженное накопление GM2 (рис. 6g, h, i и расширенные данные рис. 8g). Нейроны у трансплантированных животных также восстановили активность Hex и иммунореактивность к HEXB, подтверждая, что микроглия является достаточным внешним источником (рис. 6j, k). Важно отметить, что без опосредованного BLZ945 истощения ниши трансплантация сама по себе оказывала лишь незначительное влияние на исход заболевания, что указывает на необходимость наличия пустой ниши микроглии (рис. 6b–e). Действительно, улучшение поведения значительно коррелировало с долей привитых
Hexb-компетентных микроглиоподобных клеток (MLC) (рис. 6f). Следует отметить, что донорские MLC проявляли приблизительно 60% (61,49 ± 2,39%) экспрессии мРНК
Hexb и 70% (70,72 ± 0,98%) ферментативной активности Hex по сравнению с остаточной эндогенной микроглией (рис. 6l и расширенные данные рис. 8h). Более того, начало лечения в неонатальный период еще больше улучшает результаты (расширенные данные, рис. 8i–l), что в совокупности подчеркивает терапевтический потенциал раннего вмешательства и генетически улучшенных донорских клеток.
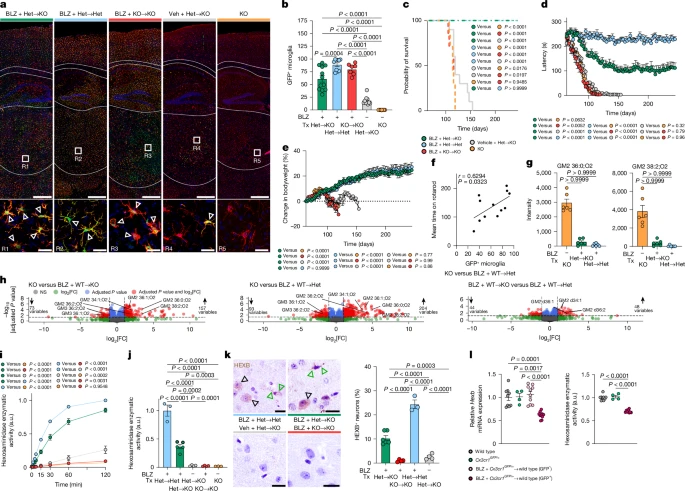 Рис. 6. Экспрессия Hexb MLC, полученными из костного мозга, предотвращает летальный фенотип ЦНС и восстанавливает гомеостаз головного мозга.
Рис. 6. Экспрессия Hexb MLC, полученными из костного мозга, предотвращает летальный фенотип ЦНС и восстанавливает гомеостаз головного мозга.
Для анализа транскрипционных изменений мы выполнили секвенирование snRNA ядер таламуса от трансплантированных и нетрансплантированных мышей Hexb-/- и Hexb+/- (Расширенные данные на рис. 8m–o). Неконтролируемая кластеризация выявила три основных кластера микроглии: c1 был обогащен гомеостатическими генами (Tmem119, P2ry12 и Sall1), c2 показал повышенную экспрессию генов, ассоциированных с заболеванием (Spp1, Gpnmb, Ctsb и Cst7), а c0 состоял в основном из MLC донорского происхождения с отчетливым профилем (расширенные данные, рис. 8p).–т). Микроглия хозяина в трансплантированном Hexb-/- мозге показала снижение активности c2 и повышенную экспрессию гомеостатических генов (расширенные данные, рис. 8u, v), что свидетельствует о частичной нормализации транскриптома посредством приживления MLC. В целом, эти данные убедительно свидетельствуют о том, что полученные от доноров MLC восстанавливают лизосомальную функцию, ограничивают накопление GM2 и помогают восстановить гомеостатическое состояние микроглии, предлагая клинически применимую стратегию лечения болезни Сандхоффа.
Shared disease pattern in Sandhoff disease brains
Чтобы сравнить наши результаты у мышей с заболеванием человека, мы провели гистопатологический анализ тканей ЦНС пациентов с болезнью Сандхоффа и контрольной группы, сопоставимой по возрасту и полу (дополнительная таблица 1). Иммуногистохимия IBA1 показала наличие крупной пенистой микроглии с втянутыми отростками только в образцах с болезнью Сандхоффа (рис. 7а), особенно богатой в мозжечке (расширенные данные рис. 9а). Эти микроглии показали повышенные уровни KIM1P, p22phox, лизоцима и LAMP2, что согласуется с активацией лизосом, наблюдаемой у мышей (расширенные данные на рис. 9b). Окрашивание гематоксилином и эозином показало избыточное количество внутриклеточных включений и гиперцеллюлярных пространств Вирхова–Робина (расширенные данные, рис. 9в, г). Включения были luxol fast blue (LFB)+ и в основном обнаруживались в нейронах и микроглии (рис. 7а). Как и у мышей, микроглиоз у индивидуумов с болезнью Сандхоффа сопровождался отеком аксонов и повреждением ядер таламуса (рис. 7а) с уменьшением плотности аксонов в белом веществе таламуса (расширенные данные рис. 9е). Иммуногистохимия SMI31 показала накопление фосфорилированных нейрофиламентов в телах нейронов, тогда как SMI35 и SMI312 показали увеличение аксонов и нарушение транспорта аксонов (расширенные данные, рис. 9f).
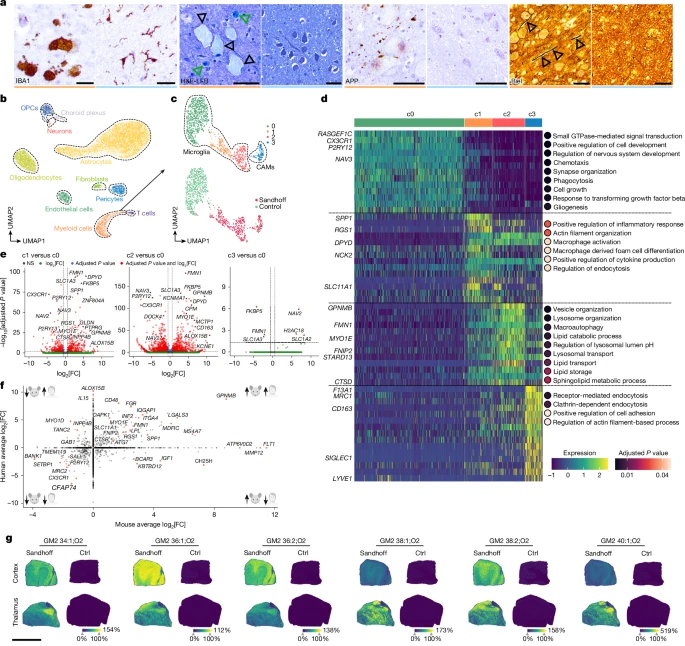 Рис. 7. Фенотип микроглии у пациентов с болезнью Сандхоффа отражает признаки заболевания, наблюдаемые у мышей Hexb-/-.
Рис. 7. Фенотип микроглии у пациентов с болезнью Сандхоффа отражает признаки заболевания, наблюдаемые у мышей Hexb-/-.
Затем мы транскрипционно охарактеризовали микроглию с помощью snRNA-seq на 14 657 единичных ядрах таламических образцов. Аннотация к типу клеток идентифицировала различные структурные типы клеток ЦНС, включая астроциты, олигодендроциты, иммунные клетки, клетки-предшественники олигодендроцитов и нейроны, среди прочих (рис. 7b и расширенные данные рис. 9g). Подсистема и повторная кластеризация выявили четыре кластера миелоидных клеток (рис. 7в), c0-2 – микроглии (CX3CR1, P2RY12, NAV3) и c3 - CAMs (F13A1, MRC1, CD163, LYVE1) (рис. 7d). Гомеостатическая микроглия была сосредоточена в c0, тогда как клетки, связанные с заболеванием, появились в c1 и c2 (рис. 7c и расширенные данные на рис. 9h). Они сильно снижали уровень CX3CR1, P2RY12, TMEM119 и SALL1 и повышали уровень GPNMB, MS4A7, MYO1E, SPP1 и LPL, отражая реакцию мыши (рис. 7d–f и расширенные данные рис. 9i). Анализ GO также показал усиление воспалительных сигнальных путей (c1) и лизосомальных путей/путей аутофагии (c2) (рис. 7d). В конечном счете, нецелевая липидомика и пространственный анализ ткани мозга пациента показали наличие видов GM2 — таких как GM2 34:1; O2 до 40:1; O2 в коре головного мозга и таламусе, что полностью соответствует профилю GM2 мыши (рис. 7g и расширенные данные рис. 9j, k). В заключение, мозг пациентов с болезнью Сандхоффа воспроизводит транскрипционные, гистологические и липидомные признаки, наблюдаемые у мышей с дефицитом Hexb, подтверждая общий механизм заболевания.
Discussion
Наше исследование описывает связь между микроглией и нейронами, которая обеспечивает нормальный гомеостаз мозга (расширенные данные на рис. 10). Сочетая объективную липидомику, одноклеточную транскриптомику, пространственную визуализацию липидов и новые генетические модели, мы определили функциональную взаимосвязь, при которой лизосомальный фермент микроглии Hex регулирует липидный баланс нейронов. Мы начали с мониторинга экспрессии основного гена микроглии на моделях нейродегенерации и демиелинизации. Хотя экспрессия Tmem119, Siglech и Sall1 была снижена, Cx3cr1 и Hexb оставались стабильными. Используя мышей-репортеров HexbtdT23, мы подтвердили микроглиальную специфичность и регуляцию развития Hexb. Хотя нейроны экспрессировали следы мРНК Hexb (примерно в 200 раз ниже), ее функциональное значение было неясно24.
Глобальная делеция Hexb вызывала раннее начало нейродегенерации, что согласуется с болезнью Сандхоффа3,25 у человека. Однако лежащие в основе молекулярные механизмы и задействованные клеточные пути оставались неполными. Было описано, что дисфункциональный Hex приводит к накоплению ганглиозидов в нейронах и последующей гибели нейрональных клеток, а реактивная микроглия способствует сохранению нейротоксической среды33. Наши гистологические и одноклеточные данные показывают, что активация микроглии предшествует изменениям астроцитов или нейронов, что позволяет предположить, что микроглия реагирует на них раньше других. Наряду с нашим выводом о том, что полная замена микроглии останавливает прогрессирование заболевания, это подтверждает классификацию дефицита Hexb как новой микроглиопатии36.
Транскрипционный профиль Hexb-/-микроглии частично напоминал активированные состояния, наблюдаемые в других моделях нейродегенерации, с повышением регуляции Apoe, Ctsb, Csf1, Igf1, Lyst и Gpnmb (ссылки 27,37), наряду с различными маркерами, такими как Apobec1, Flt1, Colec12, Adam33 или Atp6v0d2. Gpnmb, наиболее активируемый ген, кодирует трансмембранный гликопротеин, индуцируемый в насыщенных липидами макрофагах38,39 и связанный с противовоспалительными свойствами40 и восстановлением тканей41. Важно отметить, что пути фагоцитоза и аутофагии, которые обычно нарушаются при различных нарушениях накопления лизосом [42], были сильно изменены в Hexb-/- микроглии, что подчеркивает ключевую роль фермента в функционировании лизосом.
Другим сильно активируемым геном в микроглии с дефицитом Hexb был Ms4a7, который ранее считался маркером периферического миелоидного происхождения43. Однако наши данные свидетельствуют о том, что Ms4a7 отражает состояние активации, а не онтогенез, что подчеркивает ограничения использования одногенных маркеров для различения резидентных и периферических миелоидных популяций в ЦНС.
Чтобы понять, как потеря фермента микроглии вызывает нейродегенерацию, мы проанализировали механизм переноса Hex. Микроглия выделяет фермент по пути Гольджи во внеклеточное пространство, где он поглощается нейронами для расщепления ганглиозидов GM2. Липидомика подтвердила, что GM2 является наиболее избыточно накапливаемым липидом. Примечательно, что количество видов GM2 с более длинными церамидными каркасами увеличивалось по мере прогрессирования заболевания, что отражает изменения в составе ганглиозидов в постнатальном развитии44. До сих пор неизвестно, как происходит этот сдвиг и какое функциональное значение он имеет для гомеостаза и болезни Сандхоффа.
Накопление GM2 в мозге мышей и человека с дефицитом Hexb коррелирует с выраженным микроглиозом, что позволяет предположить причинно-следственную связь между этими двумя явлениями. Действительно, GM2 — но не GM1 или GM3 — индуцировал провоспалительные цитокины в микроглии, что указывает на специфический иммунный ответ. Интересно, что, хотя ганглиозидоз GM1 также связан с выраженной активацией микроглии и высвобождением цитокинов45, этот эффект, по-видимому, не связан с прямым распознаванием GM1. На самом деле, было показано, что GM1 оказывает противовоспалительное действие на микроглию46. Мы определили MGL2 как ключевой рецептор, опосредующий эффекты GM2. Экспрессируемый в дендритных клетках, макрофагах и микроглии, MGL2 связывает терминальные остатки GalNAc30. В целом, его передача сигналов зависит от контекста и лигандов47, что свидетельствует как о про-, так и о противовоспалительном ответе48,49.
Чтобы точно определить типы клеток, вызывающих заболевание, in vivo, мы создали условные нокауты, нацеленные на Hexb в определенных компартментах. Вопреки нашим ожиданиям, удаление Hexb в микроглии само по себе не вызывало заболевания. Только совместное истощение нейронов и микроглии воспроизводило полный фенотип, указывая на функциональную избыточность. Аналогичная компенсация наблюдалась и в других случаях — например, делеция Grn в микроглии сама по себе не вызывает патологии ЦНС50. В целом, хотя отсутствие функционального гена Hexb в микроглии само по себе не вызывает нейродегенерации, присутствия функционального гена Hexb только в микроглии достаточно для предотвращения нейродегенерации. Однако микроглия, вероятно, вносит свой вклад в патологию, усиливая воспаление. Мы описали высвобождение IL-6, TNF, CCL3, CCL4 и CCL17 из микроглии при воздействии GM2 и выявили повышенную регуляцию фагоцитарных путей в микроглии, потенциально способствующую прогрессированию заболевания. Аналогичный механизм был описан при болезни Гоше, при которой накапливающая липиды микроглия фагоцитирует живые нейроны51, и, в более широком смысле, нарушение липидного обмена связано с нейровоспалением52,53.
Помимо GM2, мы идентифицировали несколько липидов, накапливающихся в мозге мышей–мутантов Hexb, включая GA2, GalNAc–GM1 36:1-O2 (также известный как asialo-GM2) и BMP 44:12. Наша пространственно-временная липидная карта выявила патологические очаги, но прямая связь между накоплением ганглиозидов и потерей нейронов остается неясной. Недавнее исследование показало, что присущая нейронам передача сигналов cGAS-STING приводит к нейродегенерации, связанной с Hexb54, но специфическая уязвимость подтипов нейронов к стрессу GM2 требует дальнейшего изучения.
В настоящее время не существует методов лечения болезни Сандхоффа. Помимо симптоматического лечения, доступные и одобренные в настоящее время методы лечения нарушений накопления лизосом — заместительная ферментная терапия, субстратредуцирующая терапия или шаперонная терапия55 — часто являются непомерно дорогими и в первую очередь служат для замедления прогрессирования заболевания, а не для его остановки. Примечательно, что заместительная ферментная терапия не может эффективно бороться с поражением ЦНС, наблюдаемым при многих нарушениях накопления лизосомами , поскольку гематоэнцефалический барьер препятствует попаданию вводимых ферментов в паренхиму головного мозга. Генная терапия, однако, многообещающа: введение комплементарной ДНК Hexb на основе AAV показало успех у мышей56, и эти результаты были перенесены на ганглиозидоз GM2 человека57, но достижение полного охвата ЦНС остается сложной задачей. Трансплантация костного мозга предлагает альтернативу, но показала ограниченную пользу при ганглиозидозе gm258,59, вероятно, из-за недостаточного приживления миелоидных клеток, компетентных к ферментам. Предыдущие исследования, включая наше, показали, что для приживления миелоидных клеток требуется предварительное кондиционирование ЦНС (например, облучение)60, которое может быть значительно усилено в нескольких моделях воспаления и нейродегенерации 61,62. Тем не менее, замещение микроглии редко превышает 30%. Однако недавно у пожилых пациентов без заболеваний ЦНС был описан более высокий обмен микроглии и CAM с циркулирующими клетками крови [63]. В исследовании, проведенном на мышиной модели болезни Сандхоффа, было улучшено приживление клеток Hexb+ MLC путем сначала фармакологического уничтожения резидентной микроглии, а затем трансплантации культивированной мышиной микроглии в открытую нишу, что предотвратило симптомы заболевания и ослабило нейродегенерацию64. Основываясь на этих результатах, мы использовали клинически применимую трансплантацию костного мозга в сочетании с истощением микроглии для эффективной замены микроглии и продемонстрировали, что донорские клетки восстанавливают ферментативную активность, снижают GM2 и сдвигают транскрипцию микроглии в сторону гомеостаза. Раннее вмешательство еще больше улучшило результаты. Этот подход к лечению также может стать новым потенциальным терапевтическим вариантом для лечения ганглиозидозов человека, опосредованных микроглией. Аналогичным образом, наше детальное изучение перекрестных связей между микроглией и нейронами, которые регулируют оборот GM2, может послужить основой для разработки стратегий клеточной заместительной терапии при болезни Сандхоффа и других ганглиозидозах. Таким образом, наши результаты расширяют понимание функций микроглии в здоровой ЦНС, включая гомеостатическую регуляцию компонентов нейрональных мембран.
