Гены, белки и мутации
Cystic Fibrosis Transport Regulator

Cystic Fibrosis transport regulator protein (CFTR)
CF—cystic fibrosis;
CFTR—CF transmembrane conductance regulator;
ENaC—epithelial sodium channel;
ORCC—outwardly rectifying chloride channel;
pS—picoSiemens.
Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA.
Science 1989, 245: 1066–1073.
Generation of cAMP-activated chloride currents by expression of CFTR.
Science 1991, 251: 679–682.
Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR).
Cell 1992, 68: 809–818
Transmembrane mutations alter the channel characteristics of the cystic fibrosis transmembrane conductance regulator expressed in Xenopus oocytes.
Cell Physiol Biochem 1995, 362: 160–164.
The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity.
J Biol Chem 1995, 270: 1711–1717.
Phosphorylation by the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel.
Cell 1991, 66: 1027–1036.
Defective regulation of outwardly rectifying Cl-channels by protein kinase A corrected by insertion of CFTR.
Nature 1992, 358: 581–584.
Outwardly rectifying chloride channels in lymphocytes.
J Membr Biol 1992, 127: 49–56.
Two types of chloride channels on duct cells cultured from human fetal pancreas.
Am J Physiol 1989, 257: C240–C251.
Regulation of the gating of the cystic fibrosis transmembrane conductance regulator Cl channels by phosphorylation and ATP hydrolysis.
Proc Natl Acad Sci USA 1994, 91: 4698–4702.
Both the wild type and a functional isoform of CFTR are expressed in the kidney.
Am J Physiol 1996, 270: F1038–F1048.
Cystic fibrosis: molecular biology and therapeutic implications.
Science 1992, 256: 774–779.
A cluster of cystic fibrosis mutations in the first nucleotide-binding fold of the cystic fibrosis conductance regulator protein.
Nature 1990, 346: 366–369.
Abnormal localization of the cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia.
J Cell Biol 1992, 118: 551–559.
Maturation and function of the cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2.
Mol Cell Biol 1991, 11: 3886–3893.
Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene.
Proc Natl Acad Sci USA 1990, 87: 8447–8451
Mutations in CFTR associated with mild disease for Cl-
 channels with altered pore properties.
channels with altered pore properties.Nature 1993, 362: 160–164.
CFTR as a cAMP dependent regulator of sodium channels.
Science 1995, 269: 847–849.
CFTR and outward rectifying chloride channels are distinct proteins with a regulatory relationship.
Nature 1993, 363: 263–268.
Cystic fibrosis transmembrane conductance regulator is required for protein kinase A activation of an outwardly rectified anion channel purified from bovine tracheal epithelia.
J Biol Chem 1995, 270: 1521–1528.
The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel.
J Biol Chem 1994, 269: 20584–20591.
The multidrug resistance (mdr1) gene product functions as an ATP channel.
Proc Natl Acad Sci USA 1993, 90: 312–316.
Cystic fibrosis transmembrane conductance regulator associated ATP and adenosine 3-phosphate 5-phosphosulfate channels in the endoplasmic reticulum and plasma membranes.
J Biol Chem 1996, 272: 7746–7751
Failure of the cystic fibrosis transmembrane conductance regulator to conduct ATP.
Science 1996, 271: 1876–1879.
CFTR channels expressed in CHO cells do not have detectable ATP conductance.
J Membr Biol 1996, 151: 139–148.
Purified cystic fibrosis transmembrane conductance regulator (CFTR) does not function as an ATP channel.
J Biol Chem 1995, 271: 11623–11626.
CFTR regulates outwardly rectifying chloride currents through an autocrine mechanism involving ATP.
Cell 1995, 81: 1063–1073.
Cystic fibrosis transmembrane conductance regulator and adenosine triphosphate.
Science 1997, 275: 1324–1325.
Cystic fibrosis hetero-
and homozygosity is associated with inhibition of breast cancer growth.Nat Med 1996, 2: 593–596.
Mutant (delta F508) cystic fibrosis transmembrane conductance regulator Cl-
channel is functional when retained in endoplasmic reticulum of mammalian cells.J Biol Chem 1995, 270: 12347–12350.
Increased sulfation of glycoconjugates by cultured nasal epithelial cells from patients with cystic fibrosis.
J Clin Invest 1989, 84: 68–72.
Alteration of sulfation of glycoconjugates, but not sulfate transport and intracellular inorganic sulfate content in cystic fibrosis airway epithelial cells.
Pediatr Res 1995, 38: 42–48.
Cystic fibrosis transmembrane conductance regulator and adenosine triphosphate.
Science 1997, 275: 1325–1326.
Intrinsic anion channel activity of the recombinant first nucleotide binding fold domain of the cystic fibrosis transmembrane regulator.
Proc Natl Acad Sci USA 1992, 89: 1539–1543.
PDZs and receptor/channel clustering: rounding up the latest suspects.
Neuron 1996, 17: 575–578.
Clustering of the Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases.
Nature 1995, 378: 85–88.
Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins.
Neuron 1996, 17: 103–113.
Nucleotide triphosphates are required to open the CFTR chloride channel.
Cell 1991, 87: 775–784.
Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis.
Cell 1990, 63: 827–834.
Chloride channels in CF: lack of activation by protein kinase C and cAMP-dependent protein kinase.
Science 1989, 244: 1351–1353.
Relative ion permeability of normal and cystic fibrosis nasal epithelium.
J Clin Invest 1983, 71: 1410–1417.
Phosphorylation of cystic fibrosis transmembrane conductance regulator.
J Biol Chem 1992, 267: 12742–12752.
The cystic fibrosis transmembrane conductance regulator chloride channel. Iodide block and permeation.
Biophys J 1992, 62: 1–4.
Phosphorylation-regulated Cl-
channel in CHO cells stably expressing the cystic fibrosis gene.Nature 1991, 352: 628–631.
Cystic fibrosis decreases the apical membrane chloride permeability of monolayers cultured from cells of tracheal epithelium.
Proc Natl Acad Sci USA 1985, 82: 6167–6171.
cAMP-inducible chloride conductance in mouse fibroblast lines stably expressing the human cystic fibrosis transmembrane conductance regulator.
Proc Natl Acad Sci USA 1991, 88: 7500–7504.
Regulation of human airway surface liquid.
Respir Physiol 1995, 99: 3–12.
Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation.
J Clin Invest 1986, 78: 1245–1252.
Bacterial periplasmic permeases belong to a family of transport proteins operating from Escherichia coli to human traffic ATPases.
FEMS Microbiol Rev 1990, 75: 429–446.
MHC class II region encoding proteins related to the multidrug resistance family of transmembrane transporters.
Nature 1990, 348: 738–741
Higgins CF, Hiles ID, Salmond GPC, Gill DR, Downie JA, Evans IJ, Holland IB, Buckel SD, Bell AW, Hermondson MA:
A family of related ATP-binding subunits coupled to many distinct biological processes in bacteria.
Nature
1986,
323:
448–450.
Structural model of ATP binding proteins associated with cystic fibrosis, multidrug resistance and bacterial transport.
Nature 1990, 346: 362–365.
Distantly related sequences in the α and β subunits of ATP synthase, myosin, kinases and other ATP requiring enzymes and a common nucleotide binding fold.
EMBO J 1982, 1: 945–951.
CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR (CFTR)
Регулятор CFTR
Current Opinion in Cell Biology Vol. 9, No. 4, August 1997The cystic fibrosis transmembrane conductance regulator and ATP
Sreenivas Devidas, William B Guggino
Current Opinion in Cell Biology 1997, 9:547-552.
Введение
Quinton предположил в 1983, что транспорт хлорида нарушен при cystic fibrosis (CF), а 6 лет спустя ген CF был клонирован [1], а последующее изучение CF transmembrane conductance regulator (CFTR) показало, что хлорные канальцы обладают линейным I/V отношением (т.е. не приобретает каких-либо rectification характеристик и линейно увеличивается с повышением напряжения ) и в 9–11 picoSiemens (pS) одно-канальной проводимостью [2] [3]. Мутации в CFTR могут нарушать проводимость белка CFTR и его процессинг, а также регуляцию и способность CFTR взаимодействовать с другими ионными канальцами [4] [5] [6] [7] [8] [9] [10] [11]. Отдельные мутации обычно оказывают более одного эффекта, напр., самая распространенная δPhe508 мутация. Первичный эффект ее заключается в нарушении процессинга белка, так что большая часть белка никогда не проходит полного процессинга, не поступает в апикальные клеточные мембраны, а остается в эндоплазматическом ретикулеме [1] [12] [13] [14] [15] [16] [17].
CFTR является регулятором эпителиальных натриевых канальцев (ENaCs) [18] и внешних очищающих хлорных канальцев (ORCCs) [7] [19] [20], а также вовлечен в транспорт АТФ [21] [22] [23••] [24••] [25•] [26]. Регуляция ORCCs с помощью CFTR была подтверждена и было показана потребность в нем для АТФ в качестве аутокринного сигнала [27]. Позднее было также показано, что CFTR является негативным регулятором ENaCs [18].
The CFTR conducts chloride and ATP
Предыдущие исследования CFTR и АТФ показали, что CFTR может транспортировать АТФ. Cantiello с авт и др. [21] [22] [28••] [29] показали, что CFTR двойственен в отношении АТФ и хлорных канальцев. Это согласуется с ранними находками[2], которые показали, что CFTR является multi-ion-selective каналом с halide избирательностью Cl->I-. На клеточном уровне diphenylamine-2-carboxylic acid (DPC, неспецифический блокатор хлорных каналов)-чувствительные хлорные каналы, активируются с помощью цАМФ. Природа этого пути тока хлоридов тщательно исследовалась. Подтверждено, что CFTR является хлорным каналом. Показано, что [21] цАМФ-стимулированные АТФ токи м.б. измерены только в CFTR-трансфицированных клетках. Подтверждено, что АТФ, действительно, несет заряд. Сходные результаты получены Schwiebert et al. [27]. Эти авторы предполагают [27] что CFTR или непосредственно транспортирует АТФ или регулирует активность тесно-ассоциированных АТФ-высвобождающих каналов или регулирует exocytotic высвобождение АТФ в результате наложения осмотического градиента хлора на внутриклеточные пузырьки, нагруженные АТФ.
Pasyk and Foskett [30] показали, что CFTR ассоциирован с регулируемым проведением АТФ. Кроме того CFTR проницаемы также и для других адениновых нуклеотидов, adenosine-3-phosphate-5' phosphosulfate, универсального донора sulfate. Авт. полагают, что отсутствие такого проведения , обусловленно отсутствием нормального гликопротеина и нарушением сульфатации у пациентов с CF [31] [32]. Транспорт хлора и АТФ м. зависеть от типа клеток.
Две группы исследователей(Abraham et al. [21] [22] [28••] [29] and Schwiebert et al. [27]) описали сходное АТФ высвобождение([28••]; EM Schwiebert, personal communication). Большинство нормальных или CFTR-экспрессирующих клеток отвечает на цАМФ стимуляцию высвобождением АТФ во внеклеточную среду. Нетрансфицированные или CF типы клеток не высвобождают АТФ в ответ на цАМФ стимуляцию.
Abraham et al. [28••] показали, что антисмысловые олигонуклеотиды CFTR прекращают высвобождение АТФ.
The CFTR conducts chloride but not ATP
Reddy et al. [24••] изучая CFTR-опосредованное высвобождение АТФ показали, что АТФ не экспортируется через CFTR в апикальные мембраны потовых протоков. Кроме того на клетках Calu-3 (a human lung cell line) показали, что в них CFTR единственные хлорные каналы в апикальной мембране. Даже при сильном поляризующем потенциале не удалось выявить ток АТФ в таких клетках. Такие же результаты получены с CHO клетками, стабильно экспрессирующими CFTR [25•]. Сходные результаты получены и при использовании липидного двуслоя с рекомбинантными CFTR. Авт. пришли к выводу об отсутствии транспорта АТФ через CFTR, что согласуется с размерами пор CFTR (<5.5 Å) тогда как наименьший резмер АТФ (<10.5 Å). Canhui et al. [26] получили сходные результаты с очищенными CFTR.
Model for the release and function of ATP
Каковы же все-таки взаимоотношения между CFTR и АТФ — транспортирует ли CFTR или регулирует транспорт АТФ? И как АТФ, высвободившись, помогает CFTR выполнять его различные регуляторные функции?
Имеется три возможные модели длдя объяснения взаимоотношений между CFTR и высвобождением АТФ. Первая, что АТФ может проходить через сам CFTR канал(Fig. 1a). CFTR м.б. ассоциирован с мембраной и м. позволять проходить АТФ на клеточную поверхность. Имеются доказательства, что first nucleotide-binding fold (NBD1) CFTR м.б. ассоциирована с мембраной [34] а этот домен имеет β-barrel структуру, позволяющую проходить АТФ. Во-вторых, АТФ м. высвобождаться через второй независимый канал(Fig. 1b), который м. регулироваться с помощью CFTR[35•]. Напр., ко-экспрессия PSD-95 (postsynaptic density protein-95, a PDZ domain containing protein) с voltage-gated K+ каналами или N-methyl-D-aspartate (NMDA) рецепторами вызывает образорвание кластеров этих канало или рецепторов на плазменной мембране[36] [37]. В третьих, предполагается chloride-gradient-mediated, exocytotic высвобождение АТФ. Fig. 1c). Внутриклеточные пузырьки с АТФ сливаются с плазменными мембранами под действием хлоридного градиента и высвобождают АТФ с помощью экзоцитоза. Градиент хлоридов в этом случае должен задаваться CFTR.
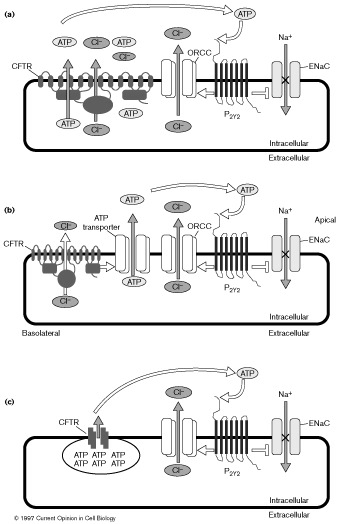 |
Figure 1 Гипотетическая модель взаимодействий между CFTR, высвобождением АТФ и канальцами, регулируемыми с помощью CFTR. The CFTR is present in airway cells; these cells are polarized and possess an apical and a basolateral surface, as shown in (b). Chloride exits the cell through the CFTR. (a) ATP as well as chloride passes through the CFTR itself. ATP may then interact with purinergic receptors such as P2Y2. These purinergic receptors, once activated by ATP, could stimulate ORCCs through second messenger pathways and thus increase chloride transport across the cell. The purinergic receptors could also inhibit ENaCs and this would result in the inhibition of sodium absorption. (b) ATP passes through a channel (the 'ATP transporter') that is separate from the CFTR but is regulated by the CFTR (indicated by the arrow between the two proteins). ATP then stimulates ORCCs and inhibits ENaCs via purinergic receptors. (c) ATP-loaded vesicles fuse at the membrane with the CFTR; this fusion is followed by an exocytotic release of ATP via a chloride osmotic gradient mediated through the CFTR. ATP then stimulates ORCCs and inhibits ENaCs via purinergic receptors. Shaded arrows indicate movement of the ions through the membrane and do not refer to movement through any particular domains of the protein concerned. |
Как высвободившийся АТФ способствует регуляторным эффектам CFTR [27] на ENaCs, ORCCs и калиевые каналы (ROMK1, на очищение АТФ-чувствительных K+ каналов)? Одним из возможных объяснений является то, что высвободившийся АТФ взаимодействует с separate purinergic рецепторами, таким как P2Y2. Рецепторы, однажды активированные с помощью АТФ, м. затем регулировать сами через пути вторичных мессенджеров. Другая возможность, что АТФ может прямо активировать ORCCs. Ингибирование ENaCs с помощью CFTR м. также управляться через прямое взаимодействие между избранными честями CFTR и ENaCs или путем ингибирования с помощью высвободившегося АТФ. Предполагается, что АТФ стимулирует purinergic рецепторы, которые стимулируют protein kinase C и тем ингибируют ENaCs путем взаимодействия протеин киназы С с α subunit of the ENaCs. Сходная гипотеза предложена для взаимодействия с ROMK1 каналами[38] [39] [40] [41] [42] [43] [44] [45] [46] [47] [48] [49] [50] [51] [52] [53].
по музыке радионяня. по производству ноутбук toshiba satellite. ноутбуки Samsung по историям