Классификация 18 Histone Deacetylases (HDACs) человека произведена по биохим., структурным и филогенетическим критериям. Базируясь на их зависимости от специфических кофакторов отделена группа I HDACs, которые являются zinc-зависимыми amidohydrolases, от группы II энзимов (известных также как класс III HDACs or SIRTs), которые нуждаются в nicotinamide adenine dinucleotide (NAD) для своей каталитической активности (Haigis and Guarente, 2006). Группа I HDACs далее подразделяется на classes I, II и IV, базируясь на их сходстве в дрожжевыми белками (Yang and Seto, 2008). В то время как class I HDACs (HDAC1,-2,-3 и -8) родственны дрожжевому транскрипционному регулятору Rpd3 (Rundlett et al., 1996; Taunton et al., 1996), энзимы class II (HDAC4,-5,-6,-7,-9,-10) обнаруживают больше сходства с yHDA1, Rpd3-паралогом (Fischer et al., 2002; Fischle et al., 2001; Fischle et al., 1999; Grozinger et al., 1999; Guardiola and Yao, 2002; Kao et al., 2000; Miska et al., 1999; Petrie et al., 2003; Tong et al., 2002; Verdel and Khochbin, 1999; Wang et al., 1999; Zhou et al., 2001; Zhou et al., 2000a). Тщательный филогенетический анализ позволил выделить HDAC11 из group I HDACs и привел к идее, что она может быть определена как член отдельного четвертого класса (Gregoretti et al., 2004).
Исходя из структуры class II HDACs произведено дальнейшее подразделение на два подкласса: class IIa (HDAC4, -5, -7 и -9) и class IIb (HDAC6 and -10) (Verdin et al., 2003). Class IIa HDACs и является предметом данного обзора и обнаруживает очень специфическую структурную организацию, которая включает два самостоятельных домена. Каталитический HDAC домен расположен в С-терминальной половине белка и соответствует HDA1-подобным последовательностям. N-терминальная часть class IIa HDACs характерна для подсемейства и содержит специфические консервативные аминокислотные мотивы, которые специализированы для связывания ряда белков, таких как ДНК-связывающие транскрипционные факторы, транскрипционные корепрессоры и шапероновые белки (Martin et al., 2007). Этот адапторный домен class IIa HDACs также содержит несколько мотивов специфически участвующих в их регуляции. Канонический nuclear localization signal обнаруживается на N-конце всех членов класса class IIa и контролирует из субклеточную локализацию (McKinsey et al., 2000a; McKinsey et al., 2001; Wang and Yang, 2001). Кроме того, несколько консервативных остатков обнаруживается по различным пост-трансляционным модификациям, таким как ubiquitination (Li et al., 2004), sumoylation (Kirsh et al., 2002; Petrie et al., 2003), фосфорилирование (Grozinger and Schreiber, 2000; Wang et al., 2000) и протеолитическое расщепление (Bakin and Jung, 2004; Li et al., 2004; Paroni et al., 2004; Scott et al., 2008).
Класс IIa HDACs, как полагают, действует как транскрипционные корепрессоры путем деацетилирования нуклеосомных гистонов вблизи промоторов мишеней. Т.к. class IIa HDACs не соединяются с ДНК, их репрессирующая активность базируется на взаимодействиях с сиквенс-специфическими ДНК связывающими белками, которые т. о., диктуют специфичность таргетинга класса IIa HDACs. Канонический пример этой модели демонстрируется взаимодействием между класса IIa HDACs и myocyte enhancer factor 2 (MEF2) транскрипционными факторами (Martin et al., 2007). Однако это простейшая модель оспаривается серией экспериментальных доказательств. Известно, что по сравнению с class I или class IIb, класса IIa HDACs являются очень неэффективными гистоновыми деацетилазами (Fischle et al., 2002; Fischle et al., 1999; Hassig et al., 1998; Hu et al., 2000). Объяснением этой прирожденной слабости активности HDAC является то, что на месте каталитического тирозинового остатка, который сильно законсервирован в др. энзимах группы I, класс IIa HDACs имеет гистидин, который компроментирует его активность в отношении acetyl-lysines (Lahm, 2007). Возникает интригующая возможность, что класс IIa HDACs может фактически не находить ацетилированные белки, но всё ещё является обнаружимым субстратом (still-to-be-discovered substrates). Кроме того, class IIa HDACs может репрессировать транскрипцию независимо от своего С-терминального каталитического домена, это указывает на то, что, по крайней мере, частично их репрессивные свойства не связаны с деацетилированием проксимальных частей гистонов (Kao et al., 2000; Lemercier et al., 2000; Sparrow et al., 1999; Wang et al., 1999; Zhou et al., 2000a). В самом деле, изолированная N-терминальная область class IIa HDACs может репрессировать транскрипцию путем рекрутирования корепрессоров, таких как HP1 или CtBP (Zhang et al., 2002; Zhang et al., 2001a; Zhang et al., 2001b). Наконец, class IIa HDACs может также моделировать транскрипционную активность своих партнеров благодаря регуляции специфических пост-транскрипционных модификаций, таких как sumoylation (Gregoire et al., 2006; Gregoire and Yang, 2005; Zhao et al., 2005) или ubiquitination (Jeon et al., 2006; Jin et al., 2004).
Помимо сходной структурной организации, class IIa HDACs также обладают одним и тем же общим механизмом регуляции, который базируется на их способности сновать между ядром и цитоплазмой в ответ на специфические внеклеточные сигналы (Martin et al., 2007). В ядре, class IIa HDACs может быть партнером транскрипционных факторов и корепрессоров, чтобы ингибировать транскрипцию. Напротив, накопление в цитоплазме class IIa HDACs делает его неспособным влиять на транскрипцию, т.к. он секвестрируется ею от гистонов и делается ферментативно неактивным как histone deacetylases (Fischle et al., 2001; Fischle et al., 2002). Многие лини доказывают, что class IIa HDAC субклеточная локализация базируется на фосфорилировании некоторых консервативных сериновых остатков, расположенных в их адапторном домене. Будучи фосфорилированными эти остатки нацеливаются на связывание с помощью 14-3-3 белков, которые способствуют CRM1-зависимому экспорту из ядра и/или цитоплазматической секвестрации class IIa HDACs (Grozinger and Schreiber, 2000; Kao et al., 2001; Liu et al., 2005; Wang et al., 2000). Напротив, мутации фосфорилируемых остатков в аланин приводят к дефициту перемещения class IIa HDACs и устраняют чувствительность к сигналу их генов мишеней (Dequiedt et al., 2003; Ha et al., 2008; McKinsey et al., 2000a; Miska et al., 2001; Wang et al., 2008; Zhang et al., 2002; Zhang et al., 2001b). Как показано для некоторых из их белков мишеней, 14-3-3 непосредственно контролируют субклеточную локализацию class IIa HDACs за счет модулирования функции их NES и NLS (Grozinger and Schreiber, 2000; McKinsey et al., 2000a; Muslin and Xing, 2000). Однако серия важных наблюдений, несовместимых с этой моделью, была упущен. Во-первых, несмотря на наличие интактных сайтов связывания 14-3-3 белков, HDAC5 мутанты, обладающие инактивирующими мутациями в их NES, остаются ядерными. Но что более важно, постоянная локализация этих NES мутантов в ядре не увеличивает их репрессивной активности. Это указывает на то, что их targeting DNA-binding партнеры скорее, чем выход из ядра, является критической ступенью при смене классом IIa HDAC транскрипционной репрессии за счет фосфорилирования (Lu et al., 2000a; McKinsey et al., 2000a). В этом контексте, 14-3-3-зависимый экспорт из ядра class IIa HDACs может служить лишь как поддерживающий механизм, который гарантирует максимальную активацию их генов мишеней (McKinsey et al., 2000a). Кроме того, некоторые данные указывают на то, что ассоциация с 14-3-3 может быть несущественно для CRM1-обеспечиваемого экспорта из ядра класса IIa HDACs (Gao et al., 2006). Полное понимание сложных механизмов, управляющих субклеточной локализацией и активностью class IIa HDAC требеует дальнеших исследований.
Итак, ингибирование фосфорилирования class IIa HDAC за счет serine-to-alanine мутаций в консенсусном 14-3-3 сайте ведет к накоплению в ядре и повышению репрессирующей активности. Это подтверждает, что модулирование фосфорилирования class IIa HDAC д. представлять уникальную возможность для контроля их биологических функций. В самом деле, активация генов мишеней для class IIa HDAC может быть достигнута за счет избыточной экспрессии протеин киназ или ингибирования протеин фосфатаз, каждое из них ведет к гиперфосфорилированию и накоплению в цитоплазме class IIa HDACs. Предпринято большое количество попыток идентифицировать протеин киназа и фосфатазы, обнаруживающие в class IIa HDAC мотивы 14-3-3 . Кстати 4 семейства serine/threonine киназ и два семейства фосфатаз участвует в этом механизме. Исторически, члены семейства Ca2+/calmodulin-dependent kinase (CaMK), особенно CaMKI и IV, были первыми киназами, которые способствовали связыванию белков 14-3-3 и экспорту из ядра class IIa HDACs. Функциональное значение этих находок было определено в различных биологических контекстах, в которых активация членов CaMK коррелировала с активацией генов мишеней для class IIa HDAC (Backs et al., 2006; Bossuyt et al., 2008; Chawla et al., 2003; Davis et al., 2003; Grozinger and Schreiber, 2000; Kao et al., 2001; Karamboulas et al., 2006b; Linseman et al., 2003; Lu et al., 2000b; McKinsey et al., 2000b). Однако эти исследования в основном базировались на избыточной экспрессии или фармакологическом ингибировании CaMK и еще предстоит оценить с помощью более определенных подходов потери функции. Недавно было показано, что protein kinase D (PKD), нижестоящий эффектор PKC, фосфорилирует сайты связывания 14-3-3 class IIa HDACs и нейтрализует их репрессивную активность (Dequiedt et al., 2005; Matthews et al., 2006; Parra et al., 2005; Vega et al., 2004a). Убедительные экспериментальные доказательства продемонстрировали, что активация PKD необходима для инактивации class IIa HDACs во время апоптоза T клеток (Dequiedt et al., 2005; Parra et al., 2005), кардиальной гипертрофии (Bossuyt et al., 2008; Vega et al., 2004a), передачи сигналов B-клеточного рецептора (Matthews et al., 2006), ремоделирования скелетных и кардиальных мышц (Fielitz et al., 2008; Kim et al., 2008a) и ангиогенеза (Ha et al., 2008; Wang et al., 2008). Мыши с редуцированной активностью CREB обнаруживают мышечную дистрофию из-за повышенной экспрессии salt-inducible kinase 1 (SIK1). Интересно, что эти мыши обнаруживали также гиперфосфорилирование HDAC5, указывая на то, что SIK1 может быть киназой class IIa HDAC. В самом деле исследования с потерей или избыточной функцией продемонстрировали, что SIK1 фосфорилирует сайты связывания 14-3-3 в HDAC5, указывая на важность CREB-SIK1-HDAC5-MEF2 оси в регуляции миогенной программы (Berdeaux et al., 2007; Takemori et al., 2009). Однако , законсервирован ли этот путь в др. MEF2-регулируемых программах остается неясным. Помимо сигналом-регулируемых протеин киназ, описанных выше, два члена семейства microtubule affinity-regulating kinase (MARK)/Par-1, которые обычно рассматриваются как постоянно активные киназы (Lizcano et al., 2004), также прямо фосфорилировали и способствовали экспорту из ядра class IIa HDACs (Chang et al., 2005; Dequiedt et al., 2006). К сожалению, несмотря на строгие биохимические данные эти исследования не получили подтверждающих доказательств, чтобы продемонстрировать биологическое значение этих находок.
Учитывая обратимую природу фосфорилирования белков, протеин фосфатазы д. быть важными как и протеин киназы в регуляции class IIa HDACs. В самом деле, Parra et al. недавно сообщили о дефосфорилировании HDAC7 с помощью миозин фосфатазного комплекса, который содержит PP1β и myosin phosphatase targeting subunit 1 (MYPT1) (Parra et al., 2007). Кроме того, результаты нашей лаб. и др. показали, что др. клеточная фосфатаза, PP2A, стабильно ассоциирует с и постоянно дефосфорилирует class IIa HDACs in vivo (Martin et al., 2008; Paroni et al., 2008). Соотв., PP2A участвует в регуляции субклеточной локализации class IIa HDAC и оказывает непосредственное влияние на их биологические функции (Illi et al., 2008; Martin et al., 2008; Paroni et al., 2008; Sucharov et al., 2006). Последовательное и/или скоординированное действие этих множественных протеин киназ и фосфатаз составлять тонко регулируемый механизм, делающий возможной соответствующую, быструю и обратимую экспрессию генов мишеней для class IIa HDAC в ответ на специфические онтогенетические сигналы.
Помимо фосфорилирования сайтов их связывания 14-3-3, class IIa HDACs является предметом дополнительных пост-трансляционных модификаций: фосфрилирования/дефосфорилирования сериновых остатков отличных от таковых в сайтаз связывания 14-3-3 (Deng et al., 2005; Paroni et al., 2008; Zhou et al., 2000b), sumoylation (Kirsh et al., 2002; Petrie et al., 2003; Tatham et al., 2001), ubiquitination (Hook et al., 2002) и расщепления каспазами (Li et al., 2004; Paroni et al., 2004; Scott et al., 2008; Martin et al., 2007; Yang and Seto, 2008).
Недавно открыты два дополнительных механизма регуляции class IIa HDACs. Во-первых, HDAC4, как было установлено, олигомеризуется с HDAC5, и в меньшей степени с HDAC9. Интересно, что в class IIa HDACs, HDAC4 является одинаково чувствительным к CaMKII-обусловленному фосфорилированию и экспорту из ядра (Backs et al., 2006). Гетеро-олигомеризация с HDAC4 приводит HDAC5 в тесную близость к CaMKII, этот делает возможным её фосфорилирование с помощью киназы и способствует её экспорту из ядра (Backs et al., 2008). Существует ли подобный механизм трансфосфорилирования и для др. class IIa HDACs и для еще не идентифицированных специфических киназ, неизвестно. Недавно сообщалось о двух исследованиях, в которых экспрессия HDAC4 регулируется на пост-транскрипционном уровне с помощью microRNAs (miRNA). Во время дифференцировки скелетных мышц, miR-1 способствует миогенезу путем доставки HDAC4 (Chen et al., 2006). Сходным образом, miR140, специфичная для хряща miRNA, может соединяться с мРНК HDAC4 и вмешивается в её трансляцию (Tuddenham et al., 2006). К сожалению, биологическое значение этих находок во время развития кости не было исследовано. Тем не менее эти важные исследования открывают новую линию изучения регуляции class IIa HDACs.
Сушествование множественных регуляторных путей, сходящихся на class IIa HDACs подчеркивает важность этих энзимов в различных биологических процессах. В последнее время исследования по генетической инактивации у мышей, проводимые в лаб. Olson оказались пригодными для выяснения биологических функций этих энзимов. Эти исследования выявили ключевую роль членов class IIa в некоторых важных процессах развития и дифференцировки. Неожиданно, несмотря на большое количество регуляторов транскрипции, доставляемых с помощью class IIa HDACs, большинство их функций, по-видимому, участвует в репрессии транскрипции MEF2 транскрипционных факторов (Fig. 1).
Chondrocyte hypertrophy
HDAC4-нулевые мыши погибают во время пренатального периода из-за тяжелой задержки роста и многочисленных скелетных аномалий, которые являются результатом избыточной дифференцировки гипертрофических хондроцитов и неадекватной эндохондральной оссификации (Vega et al., 2004b). Этот характерный фенотип был первоначально приписан способности HDAC4 репрессировать Runt-related transcription factor-2 (Runx2), хорошо известный позитивный регулятор гипертрофии хондроцитов.
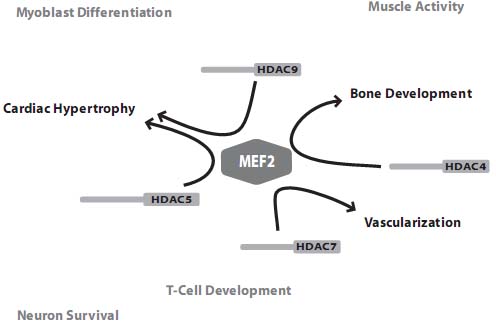 Fig. 1. Central role of MEF2 in the biological functions of class IIa HDACs. Gene targeting studies in mice have revealed tissue specific functions for each member of the class IIa HDACs. HDAC4-null mice display inappropriate chondrocyte hypertrophy whereas mice lacking HDAC7 die from cardiovascular defects and mutant mice for HDAC5 and HDAC9 develop spontaneous cardiac hypertrophy. Surprisingly, the known biological functions of class IIa HDACs have been associated with repression of the MEF2 family of transcription factors. A similar class IIa HDACsMEF2 axis could be involved in cardiac muscle development, skeletal muscle differentiation and remodelling and T-cell apoptosis.
Fig. 1. Central role of MEF2 in the biological functions of class IIa HDACs. Gene targeting studies in mice have revealed tissue specific functions for each member of the class IIa HDACs. HDAC4-null mice display inappropriate chondrocyte hypertrophy whereas mice lacking HDAC7 die from cardiovascular defects and mutant mice for HDAC5 and HDAC9 develop spontaneous cardiac hypertrophy. Surprisingly, the known biological functions of class IIa HDACs have been associated with repression of the MEF2 family of transcription factors. A similar class IIa HDACsMEF2 axis could be involved in cardiac muscle development, skeletal muscle differentiation and remodelling and T-cell apoptosis.
(Komori et al., 1997; Otto et al., 1997). В самом деле, HDAC4 физически ассоциирует с Runx2 и ингибирует его транскрипционные активности. Кроме того, фенотипические аномалии у HDAC4 KO мышей очень сильно напоминали таковые у мышей с эктопической экспрессией
Runx2 в прегипетртрофических хондроцитах (Takeda et al., 2001; Ueta et al., 2001). Точный механизм, с помощью которого HDAC4 ингибирует Runx2-обусловленную транскрипцию остается неясным. Оригинальные наблюдения указывают на то, что репрессия Runx2 с помощью HDAC4 может происходить независимо от её HDAC каталитической активности. Вместо этого, HDAC4 , как полагают, затрудняет Runx2 связывание с ДНК за счет прямой ассоциации с её Runt доменом (Vega et al., 2004b). Однако эта модель неспособна объяснить, почему HDAC4 мутанты, состоящие из C-терминального каталитического домена HDAC4, и т.о., лишенные Runx2 связывающей области, сохраняют значительную репрессивную активность. Недавнее наблюдение, что HDAC4 и HDAC5 деацетилируют Runx2 и Runx3 дает объяснение этому кажущемуся несоответствию (Jeon et al., 2006; Jin et al., 2004). Помимо предотвращения Runx2 от соединения с ДНК, HDAC4 может деацетилировать Runx2 и способствовать его ubiquitin-обеспечиваемой деградации. Такое двойное воздействие на Runx2 с помощью HDAC4 только и может объяснить её ключевую роль как негативного регулятора гипертрофии хондроцитов.
Эта кажущаяся удовлетворительной модель недавно поставлена под вопрос благодаря исследованиями тех же авт. , которые выявили неожиданную роль MEF2C в гипертрофии хондроцитов (Arnold et al., 2007). Это исследование сообщает, что homo- или даже гетерозиготные мутации
Mef2c ассоциируют с тяжелыми дефектами скелета, возникающими в результате снижения гипертрофии хондроцитов и оссификации эндохондральных костей. Учитывая преобладание оси class IIa HDACs-MEF2 в некоторых онтогенетических программах (see below), эта неожиданная находка открыла возможность, что дефекты костей, ассоциирующие с делецией Hdac4, должны, по крайней мере, частично вызываться за счет гиперактивации MEF2C. Подтверждением этой гипотезы является генетический антагонизм между HDAC4 и MEF2: избыточная эндохондральная оссификация, наблюдаемая у HDAC4 нулевых мышей, частично устраняется за счет делеции одного из аллелей
Mef2c. Напротив инактивация
Hdac4 в присутствии гетерозиготного аллеля
Mef2c частично восстанавливает нормальную эндохондральную оссификацию. Интересно, что экспрессия
Runx2 существенно уменьшается в эндохондральном хряще
Mef2c мутантных мышей, указывая тем самым, что некоторые из дефектов, ассоциированные с недостаточностью Mef2c, могут быть обусловлены за счет снижения активности Runx2. С помощью контролирующих ключевых транскрипционных регуляторов гипертрофии хондроцитов, теперь становится ясно, что HDAC4 играет центральную роль в контроле развития кости. Интересно, что Runx2 участвует на множественных стадиях развития кости, таких как дифференцировка остеобластов (Komori, 2008). используя модели
in vitro, HDAC7 недавно был идентифицирован как негативный регулятор дифференцировки остеобластов посредством своей Runx2 корепрессорной функции (Jensen et al., 2008). Кроме того, HDAC4 и HDAC5 , как было установлено, участвуют в репрессии
Runx2 посредством Smad3 во время дифференцировки остеобластов (Kang et al., 2005). В то время HDAC4 и -5 экспрессируются на значительных уровнях в мезенхимных клетках и остеобластах, нет доказательств для экспрессии HDAC7 в этом клеточном клоне. Кроме того, тяжелые скелетные дефекты, ассоциирующиеся с недостаточностью HDAC4, не наблюдались у др. class IIa HDACs мутантных мышей и это может указывать на специфическую роль HDAC4 как негативного регулятора развития кости. Более тщательная проверка развития кости у мышей с отсутствием class IIa HDACs д. выявить какое-либо функциональное перекрывание, которое может существовать между этими белками.
Myogenesis
Skeletal muscle differentiation
Формирование скелетных мышц использует детерминацию мультипотенциальных мезодермальных клеток предшественников мышечного клона и их пролиферацию как миобластов. После удаления митогенов пролиферирующие миобласты выходят из клеточного цикла и дифференцируются в многоядерные мышечные волокна. Миогенный процесс является результатом специфической миогенной программы с активацией сотен мышце-специфических генов и репрессией генов, ассоциированных с клеточной пролиферацией. MEF2 давно был известен как ключевой транскрипционный регулятор дифференцировки скелетных мышц. Логически функциональная ассоциация между членами семейства MEF2 и class IIa HDACs была оригинально протестирована в контексте миогенеза (Wang et al., 1999; Miska et al., 1999; Lu et al., 2000a; Lemercier et al., 2000).
In vitro, class IIa HDACs негативно регулирует мышечную дифференцировку посредством ассоциации с MEF2 и репрессии его генов мишеней (Dressel et al., 2001; Haberland et al., 2007; Lu et al., 2000b). Кроме того, ингибирующее действие class IIa HDACs может быть преодолено с помощью миогенных сигналов, которые нарушают взаимодействия MEF2-HDAC и стимулируют экспорт из ядра этих транскрипционных репрессоров во время дифференцировки мышц (McKinsey et al., 2002). Неожиданно нормальная повсеместная скелетная мышечная дифференцировка сохраняется у мышей, мутантных по каждой из индивидуальных class IIa HDAC. Это очевидное расхождение может быть результатом частичного функционального перекрывания, которое может существовать между членами class IIa HDAC. Однако некоторые наблюдения указывают на то, что функциональное перекрывание, наблюдаемое между class IIa HDACs
in vitro может и не существовать
in vivo. В самом деле, члены class IIa обнаруживают разные субклеточные локализации в мышечных клетках. HDAC4 в основном цитоплазматический в недифференцированных миобластах и накапливается в ядре после дифференцировки в мышечные трубки (Miska et al., 2001). Напротив, HDAC5 и HDAC7 перемещаются из ядра в цитоплазму, когда миобласты дифференцируются в мышечные трубки (Dressel et al., 2001; McKinsey et al., 2000a). Эти находки указывают на то, что class IIa HDACs реагируют по-разному на физиологические стимулы и могут таким образом выполнять разные роли во время дифференцировки скелетных мышц.
Skeletal muscle remodelling
Скелетные мышцы взрослых позвоночных состоят из типа I и типа II миофибрилл, которые отличаются в отношении размера, метаболизма и контрактильной функции. Медленно сокращающиеся или type I миофибриллы обладают окисдативным метаболизмом и резистентны к утомлению, тогда как напротив, быстро сокращающиеся или type II волокна используют гликолитический метаболизм, быстро устают и участвуют в быстрых взрывах активности. Многочисленные стимулы модулируют скелетно-мышечный фенотип и индуцируют переключение с одного типа специализированных миофибрилл на др. MEF2 преимущественно активируется в медленных, оксидативных миофибриллах (Wu et al., 2000) и активирует генную программу медленных мышечных волокон (Potthoff et al., 2007). Логически, class IIa HDACs как недавно было показано, участвует в регуляции качественных особенностей мышечных волокон. В медленных или оксидативных волокнах class IIa HDACs избирательно деградирует посредством зависимого от протеосом пути. Эта специфическая деградация class IIa HDACs смягчает их репрессию на MEF2, и возможно на др. не идентифицированные транскрипционные факторы, делая возможной индукцию генной программы. специфической для медленных скелетных мышц. Мыши, лишенные индивидуальных class IIa HDACs в своих скелетных мышцах не обнаруживают аномалий в переключении между типами волокон. Однако делеция в любой комбинаци из 4-х аллелей Hdac4, -5 или -9 приводит к усилению экспрессии генов медленных волокон и повышению процента медленных мышечных волокон, указывая на функциональное перекрывание между членами class IIa. Интересно, что избыточная экспрессия постоянно активной формы PKD и CaMKIV, во взрослых гликолтических волокнах трансгенных мышей приводит к увеличению количества медленых волокон и усиливает оксидативную способность мышц (Kim et al., 2008a; Wu et al., 2002). Предыдущие исследования показали, что эти киназы стимулируют активность MEF2 , способствуя фосфорилированию и экспорту class II HDACs из ядра (Martin et al., 2007), эти наблюдения ешё больше подтверждают роль class IIa HDACs в спецификации типа волокон.
В ответ на различные внешнесредовые и функциональные стимулы скелетные мышцы алаптируются благодаря ремоделированию биохимических, морфологических и физиологических состояний индивидуальных мышечных волокон (Bassel-Duby and Olson, 2006). Эти изменения связаны с активацией путей внутриклеточной передачи сигналов и соотв. генетического репрограммирования, приводящих к изменениями мышечной массы, контрактильных свойств и метаболических состояний. Многочисленные стимулы, такие как упражнения, электрическая стимуляция или микрогравитация могут индуцировать ремоделирование мышц. Индуцируемая нейронами электрическая активность, как известно, индуцирует специфической транскрипционное репрограммирование мышц и идентифицированы некоторые чувствительные к активности нейронов гены. Типичным примером является nicotinic acetylcholine receptors (AChRs), чья экспрессия высоко чувствительна к мышечной иннервации. Экспрессия AChR базируется на basic helix-loop-helix (bHLH) миогенном транскрипционном факторе myogenin. После иннервации индуцируемая нейронами электрическая активность вызывает активную транскрипционную репрессию myogenin по всей мышце, это совпадает со снижением уровней вне-синаптической экспрессии AChR. Недавно показано, что class IIa HDACs непосредственно участвует в регуляции экспрессии myogenin. В иннервированных мышцах, MITR, не каталитическая изоформаHDAC9, экспрессируется на высоком уровне и репрессирует транскрипцию myogenin посредством ингибирования MEF2. Напротив, денервация вызывает драматическое снижение транскрипции MITR, это коррелирует с последующей экспрессией myogenin и AChR expression (Mejat et al., 2005). Интересно наблюдение, что у MITR мутантного белка, лишенного MEF2-взаимодействующего домена, сохраняется 50% ингибирующей активности в отношении экспрессии AChR и myogenin, это указывает на то, что MITR может быть дополнительной мишенью помимо MEF2. HDAC9-нулевые мыши не обнаруживают каких-либо очевидных нарушений в функции скелетных мышц в нормальных условиях, но они сенсибилизированы к денервации. Этот фенотип сенсибилизации напоминает кардиальный фенотип, обнаруживаемый у HDAC5 и HDAC9-нулевых мышей и показывает, что класса IIa HDACs являются общими сенсорами и интеграторами стрессов.
Помимо MEF2, Dash2, Dachschund родственный транскрипционный корепрессор, чья экспрессия снижается после денервации, как сообщается, ингибирует экспрессию myogenin в иннервированных мышцах (Tang and Goldman, 2006). В противоположность MITR/HDAC9, экспрессия
HDAC4 обнаруживается на высоком уровне в ответ на денервацию, указывая тем самым, что оба class IIa HDACs могут обнаруживать противоположные эффекты на вызванную денервацией экспрессию nAChR (Cohen et al., 2007). Эта модель подтверждается наблюдением, что нейрональные импульсы также индуцируют накопление в ядре HDAC4 (Liu et al., 2005). В ядре HDAC4 ассоциирует с промотором Dach2 и тем самым активирует каскад myogenin/AChR благодая активному подавлению Dach2. Неожиданно, противоположная регуляция HDAC4 и MITR с помощью нервной активности ведет к одному и тому же результату: индукции экспрессии myogenin/AChR в результате денервации. Интересно, что эти два члена class IIa д. препятствовать репрограммированию транскрипции в зависимости от нейрональной активности в мышцах посредством неперекрывающихся, комплементарных путей. Существует ли сходная комбинаторная регуляция с помощью множественных членов class IIa также в др. транскрипционных программах с участием class IIa HDACs предстоит выяснить.
Cardiac muscle development
Формирование сердца инициируется из мезодермы эмбрионов позвоночных. На первой стадии кардиомиогенеза происходит формирование кардиомиобластов, которые экспрессируют определенный субнабор транскрипционных факторов, включая MEF2C, гомеобоксный транскрипционный фактор Nkx2-5 и членов подсемейства кардиальных GATA (Zaffran and Frasch, 2002). Эти факторы активируют экспрессию специфичных для кардиальной мышцы генов, чтобы сформировать дифференцированные кардиомиоциты. Избыточная экспрессия HDAC4 или ингибирование инактивирующей class IIa HDAC киназы CaMK, подавляет переход от мезодермы в кардиомиобласты во время кардиомиогенеза в P19 клетках (Karamboulas et al., 2006b). Неожиданно, мыши, мутантные по индивидуальным класса IIa HDACs, не обнаруживали видимых аномалий сердца. Напротив, HDAC5/9 двойные мутантные мыши склонны к перинатальной гибели из за дефектов межжелудочковой перегородки и истонченных стенок желудочков, обычно возникающих в результате аномалий роста и созревания кардиомиоцитов (Chang et al., 2004). В то время как молекулярные механизмы, лежащие в основе роли class IIa HDACs в этом процессе остаются неясными, исследования на мышах и
Drosophila выявили важность MEF2 для контроля дифференцировки кардиальной мышцы (Bour et al., 1995; Karamboulas et al., 2006a; Lilly et al., 1995; Lin et al., 1997; Ranganayakulu et al., 1995). Возможно. что, по крайней мере, частично дефекты кардиомиоцитов ассоциируют с инактивацией class IIa HDACs, возникающей в результате суперактивации транскрипционной активности MEF2. Однако возможно, что class IIa HDACs прямо или косвенно репрессируют др. транскрипционные факторы, которые важны для кардиомиогенеза. такие как SRF, CAMTA, Nkx2-5, myocardin и GATA факторы (Davis et al., 2003; Han et al., 2006; Long et al., 2007; Song et al., 2006).
Cardiac hypertrophy
Кардиальная гипертрофия означает увеличение размеров кардиомиоцитов и реактивацию специфической для плода кардиальной генной программы, это адаптивная реакция сердца на различные стрессовые воздействия. Индуцированная стрессами гипертрофия может первоначально нормализовать стрессы стенок желудочков, но избыточный гипертрофический рост сердца часто ведет к неадекватным изменениям, которые в конечном итоге ослабляют работу сердца и ведут к сердечной недостаточности (Backs and Olson, 2006; Hill and Olson, 2008).
Транскрипты для каждой class IIa HDACs, включая сплайс HDAC9 вариант MITR, обнаруживаются на высоком уровне в сердце мышей (Zhang et al., 2002). Кроме того, эктопическая экспрессия конституитивно репрессивных мутантов HDAC4, -5 и -9 (т.е. мутантов нечувствительных к signal-responsive class IIa HDAC киназам) подавляет гипертрофию первичных кардиомиоцитов и экспрессию плодных кардиальных генов in vitro (Backs et al., 2006; Vega et al., 2004a; Zhang et al., 2002). Исследования по инактивации генов у мышей предоставили доказательства важности class IIa HDACs как чувствительных к сигналам супрессоров постнатального кардиального роста. HDAC5 или HDAC9-KO мыши не дают доказательств кардиальных аномалий во время раннего постнатального периода. Однако в течение их первго года жизни у этих мутантных мышей развивается спонтанная кардиальная гипертрофия, которая, по-видимому, появляется в результате формирования гиперчувствительности к связанным с возрастом кардиальным стрессам. HDAC5 и HDAC9 мутантные мыши также усиливают гипертрофическую реакцию на повышение кровяного давления или конституитивную активацию calcineurin (Chang et al., 2004; Zhang et al., 2002). Удивительно сходные кардиальные фенотипы наблюдаются у HDAC5 или HDAC9 мутантных мышей, указывая тем самым, что эти два члена class IIa выполняют перекрывающиеся роли в контроле одних и тех же путей передачи патологических гипертрофических сигналов. К сожалению, мыши, лишенные или HDAC4 или HDAC7 нежизнеспособны (Chang et al., 2006; Vega et al., 2004b). Необходимы исследования по условным делециям генов, чтобы тестировать, играют ли HDAC4 и HDAC7 функциональные роли сходные с теми, которые HDAC5 и HDAC9 играют в кардиальной патологической реакции.
MEF2, и особенно MEF2D играют важную роль в качестве интегратора и медиатора зависимого от стресса ремоделирования взрослого сердца (Kim et al., 2008b; Lu et al., 2000a; Nadruz et al., 2003). В самом деле, Mefd2d-нулевые мыши обнаруживают ослабленную кардиальную гипертрофическую реакцию на повышенное кровяное давление и передачу адренергических сигналов (Kim et al., 2008b). Гипертрофический кардиальный рост HDAC5 или HDAC9 мутантных мышей коррелирует с суперактивацией MEF2, указывая тем самым, что MEF2 является критической мишенью для class IIa HDACs в специфических сигнальных путях, ведущих к кардиальной гипертрофии. В самом деле, т.к. инактивация аллеля Mef2d делает мышей резистентными к кардиальному ремоделированию в ответ на β-adrenergic стимуляцию, то HDAC5 или HDAC9-нулевые мыши отвечают нормально на стимуляцию isoproterenol (Chang et al., 2004; Kim et al., 2008b). Это интересное наблюдение может показывать, что только специфические сигнальные пути кардиальной гипертрофии используют class IIa HDAC-обусловленную регуляцию MEF2. С др. стороны, нечувствительность мышей, лишенных или HDAC5 или HDAC9 к adrenergic стимулам может означать функциональное перекрывание с др. членами класса class IIa в этом специфическом пути. Выяснение точной роли и специфических физиологических мишеней для каждой из class IIa HDAC в кардиальной гипертрофии д. помочь решить этот важный вопрос.
Neuronal survival
мРНК HDAC4 и HDAC5 очень обильна в головном мозге, это указывает на роль этого class IIa членов в нейронах (Grozinger et al., 1999). HDAC4 является преимущественно цитоплазматическим в cerebellar granule neurons (CGNs), а сигналы, способствующие её ядерной транслокации или экспрессии у конституитивных ядерных мутантов коррелируют с апоптозом (Bolger and Yao, 2005). Сходным образом, HDAC5 в основном цитоплазматическая в CGNs, культивируемых в деполяризующих условиях. Переключение CGNs на не поляризующую среду, которая вызывает транслокацию из цитоплазмы в ядро HDAC4 и -5, коррелирует с индукцией апоптоза (Linseman et al., 2003). MEF2 является хорошо известным фактором в нейронах (Mao et al., 1999) и как ожидается, индукция апоптоза с помощью HDAC4 и -5 коррелирует с репрессией транскрипционной активности MEF2 (Bolger and Yao, 2005; Linseman et al., 2003). Однако могут ли HDAC4 и/или HDAC5 находить какой-либо др. фактор помимо MEF2 остается неясным. Интересно, что HDAC4, по-видимому, выполняет более преобладающую роль в гибели нервных клеток, т.к. siRNA-обусловленное ингибирование HDAC4 эффективно супрессирует гибель нейронов, снова указывая на функциональное перекрывание между HDAC4 и HDAC5 (Bolger and Yao, 2005). Кроме того, мыши с нокдауном HDAC5 не обнаруживают очевидных дефектов головного мозга, тогда как мыши, лишенные HDAC4, имеют аномальной формы головной мозг и экзэнцефалию, возможно в результате формирования преждевременной оссификации головного мозга (Vega et al., 2004b). Однако более тщательная проверка HDAC4-нулевых мышей выявила задержку в образовании folia, указывая на способствующую выживанию роль скорее, чем про-апоптическую роль HDAC4 (Majdzadeh et al., 2008; Vega et al., 2004b). В самом деле, недавнее исследование сообщило, что HDAC4 предупреждает low-potassium индуцированную гибель нервных клеток, посредством ингибирования активности cyclin-dependent kinase-1 (CDK1). В подтверждение своих результатов авт. описали более высокую активность CDK1 в головном мозге HDAC4
-/- мышей (Majdzadeh et al., 2008). Сходным образом, исследования с использованием модели дегенерации сетчатки у мышей, показали, что HDAC4 способствует выживанию фоторецепторных клеток (Chen and Chepko, 2009). Интересно, что эта способствующая выживанию функция HDAC4 ассоциирует с её присутствием в цитоплазме и может быть независимой от её ядерной репрессивной функции. Причина этих противоречивых результатов остается неясной и нуждается в дальнейших исследованиях роли class IIa HDACs в нейронах.
Immune cells
После вступления в тимус предшественники Т клеток дифференцируются в зрелые лимфоциты вследствие сложного и высоко регулируемого процесса (Ciofani and Zuniga-Pflucker, 2007). Полное созревание развивающихся тимоцитов указывает на то, что они успешно проходят через серию онтогенетических КПП (checkpoints). После полной перестройки своих T-cell receptor (TCR) цепей, тимоциты становятся CD4++/CD8, или double-positive (DP) тимоцитами и подвергаются негативной и позитивной селекции. Во время этих процессов их TCR были протестированы в отношении способности взаимодействовать с selfpeptide bound to major histocompatibility complex (MHC) молекулами из antigen-presenting cells (APCs) тимуса. Позитивный отбор д. лишь позволить дальнейшую дифференцировку тимоцитов, несущих функциональный TCR. Потенциально аутореактивные тимоциты, несущие TCR со строгим сродством к MHC-self пептидным комплексам, делетируются с помощью апоптического процесса негативной селекции. Разные онтогенетические судьбы созревающих тимоцитов диктуются интеграцией различных сигналов и трансляцией этих сигналов в изменения генной экспрессии.
У человека HDAC7 высоко экспрессируется в тимусе, сердце и легких. В тимусе HDAC7 временно и преимущественно экспрессируется в CD4/CD8 DP тимоцитах (Dequiedt et al., 2003). В соответствии с этим, HDAC7 ассоциирует с MEF2D и репрессирует экспрессию серии генов мишеней для MEF2, участвующих в негативной и позитивной селекции, такие как про-апоптический orphan ядерный рецептор Nur77 (Dequiedt et al., 2003; Kasler and Verdin, 2007). Высвобождение от HDAC7-обусловленной репрессии достигается посредством сложного сигнального каскада. Вовлечение TCR с помощью MHCself пептидов приводит к диссоциации HDAC7-MEF2D комплексов, фосфорилированию и накоплению в цитоплазме HDAC7, это в конечном итоге ведет к дерепрессии генов мишеней для MEF2 (Dequiedt et al., 2005). Интересно, что анализ на основе микромассивов показал, что HDAC7 может регулировать как MEF2 зависимые. так и независимые гены (Kasler and Verdin, 2007). Однако как HDAC7 регулирует транскрипцию независимо от MEF2 не изучено. Среди регулируемых с помощью MEF2-HDAC7 генов в тимоцитах это исследование также идентифицировало HDAC5, это подтверждает роль этого class IIa членов во время дифференцировки Т клеток.
В В клетках передача сигналов от рецепторов В-клеточного антигена, как было показано регулирует фосфорилирование HDAC5 и HDAC7 , локализацию и репрессивную функцию (Matthews et al., 2006). Даже если химическое ингибирование HDACs подтверждает роль HDACs в B-клеточной дифференцировке и жизнеспособности, транскрипционные мишени и функциональное значение этих находок остается неизвестным.
Vascularisation
Ранние исследования с использованием общих HDAC ингибиторов указывали на возможное участие zinc-зависимых HDACs в транскрипционном контроле экспрессии эндотелиальных генов и в сосудистом развитии (Kim et al., 2001; Kwon et al., 2002). Однако вплоть до недавнего времени экспериментальные доказательства участия class I и class II HDACs в этом процессе отсутствовали.
Исследования с гибридизацией
In situ выявили специфическую экспрессию
HDAC7 в сосудистом эндотелии во время эмбриогенеза мышей. В согласии с этим инактивация гена
HDAC7 повсеместно или эндотелий специфическим образом приводила к драматическим последствиям сосудистой целостности, мутантные мыши погибали
in utero из-за расширений кровеносных сосудов, разрывов и гемморагий. Кроме того, siRNA-обусловленная инактивация HDAC7 в human umbilical vein endothelial cells (HUVECs) предупреждала образование капилляр-подобных структур
in vitro (Chang et al., 2006). Анализ микромассивов выявил, что HDAC7 регулирует экспрессию некоторых генов, кодирующих внеклеточный матрикс и адгезивные белки, среди которых секретируемая matrix metalloprotease 10 (MMP-10). Регуляция экспрессии MMP-10 с помощью HDAC7 базируется на её ассоциации с MEF2C, фактором, участвующим в развитии кровеносных сосудов и целостности сосудов (Lin et al., 1998), и составляет ещё один пример, в котором ось class IIa HDACs-MEF2 контролирует важрную генетическую программу развития. Несоотв. экспрессия MMP10 у HDAC7 нулевых мышей, можно ожидать, будет нарушать межклеточную адгезию и может таким образом объяснить дефекты, связанные с инактивацией HDAC7. Однако более недавние исследования проливают некоторый свет на роль HDAC7 во время ангиогенеза и васкуляризации. Используя полный набор
in vitro подходов, Mottet с коллегами показали, что неспособность с молчащей HDAC7 клеток HUVECs собираться в трубчато-подобные структуры
in vitro может быть результатом альтераций в их подвижности (Mottet et al., 2007). Кроме того, молчание HDAC7 коррелирует с повышенной экспрессией PDGF-B, регулятора миграции и тубулогенеза из эндотелиальных клеток (De Marchis et al., 2002). Молекулярные механизмы, лежащие в основе транскрипционного контроля PDGF-B с помощью HDAC7 неизвестны, эти находки открывают возможность, что HDAC7 может регулировать множественные стадии во время формирования васкулатуры. Интересно, что молчание HDAC7 ассоциирует также с измененной морфологией клеток, что также важно для их ангиогенных свойств (Holderfield and Hughes, 2008). Учитывая это потенциальное воздействие на множественные аспекты васкулогенеза, идентификация генов мишеней для HDAC7 становится необходимой ступенью для лучшего понимания сосудистого развития.
Future prospects
As described above, the past 5 years have witnessed the emergence of class IIa HDACs as key regulators in several important biological processes. Through their signal-responsive regulation and their ability to repress the transcriptional activity of numerous transcription factors, class IIa HDACs integrate and translate extracellular signals into specific genetic activation programs. Recent studies in mice deficient for each class IIa member have corroborated this model and revealed major defects in important developmental programs such as embryonic bone formation and vascularisation and cardiac hypertrophy (see above). While groundbreaking, these studies have focused on the functions of class IIa HDACs leading to the most spectacular phenotypical defects. However, the early observation that class IIa HDACs are expressed in multiple tissues suggests that they might have several biological functions, depending on the cellular context. More careful examination of class IIa HDAC null-mice should identify less obvious phenotypical defects and therefore suggest additional biological functions for these enzymes. For instance, while HDAC4-deficient mice die perinatally from skeletal defects, demonstrating the key role of this class IIa member in bone development, they also exhibit cerebellar abnormalities which could support a role for HDAC4 in neuronal survival (Majdzadeh et al., 2008).
Surprisingly, and despite the fact that they can associate with a plethora of partners, the biological functions of class IIa HDACs seem to rely almost uniquely on their association with MEF2 transcription factors. MEF2 has been implicated in a myriad of developmental, physiological and pathological processes, each of which could potentially be regulated by a class IIa HDACsMEF2 axis. Amongst the MEF2-regulated processes, the unique or redundant in vivo functions of class IIa members remain to be investigated in contractile myofibril assembly (Hinits and Hughes, 2007), craniofacial development (Verzi et al., 2007), postsynaptic morphogenesis and brain development (Shalizi et al., 2006; Shalizi and Bonni, 2005). Inversely, several lines of experimental evidence suggest that class IIa HDACs can regulate transcription independently of MEF2 (Kasler and Verdin, 2007). Nonetheless, whether MEF2-independent biological functions exist for class IIa HDACs remains an unanswered question and deserves to be thoroughly adressed. The existing studies about the in vivo functions of class IIa HDACs have overlooked the many other transcriptional targets of these enzymes and often linked the observed physiological defects with hyperactivation of MEF2. The existence of knock-out mice for each class IIa HDAC member gives now the opportunity to validate additional known class IIa HDAC partners besides MEF2.
Functional inactivation of class IIa HDACs via signal-dependent phosphorylation and nuclear export has been extensively illustrated in in vitro systems (Martin et al., 2007). This mechanism is now being validated in vivo by recent studies showing the importance of known class IIa HDAC kinases in cardiac hypertrophy (Backs et al., 2006; Fielitz et al., 2008; Harrison et al., 2006) and skeletal muscle function and remodeling (Kim et al., 2008a).
Logically, class IIa HDAC kinases should also be involved in other class IIa HDAC regulated genetic programs such as chondrocytes hypertrophy, neuron and immune cells apoptosis (Dequiedt et al., 2005; Parra et al., 2005), and blood vessel development (Harrison et al., 2006).
The establishment of class IIa HDACs as key modulators of various differentiation and developmental programs, especially in pathological related processes, potentially offers new therapeutic strategies for various human diseases. A still limited number of studies have reported alterations of class IIa HDACs in several cancers (Chaabouni et al., 2006; Ouaissi et al., 2008; Ozdag et al., 2006; Yuki et al., 2004) and neuronal disorders (Hoshino et al., 2003; Iga et al., 2007; Renthal et al., 2007). Armed with the new insights into their regulation and biological functions, it seems possible to specifically modulate class IIa HDAC activity and normalize pathological gene expression patterns in disorders such as cardiac hypertrophy, hearth failure, tumour growth or skeletal muscle wasting.
Сайт создан в системе
uCoz 