НАУЧНЫЕ ОБЗОРЫ витие полукружных каналов и окружающее их перилим- Мыши, дефицитные по генам НтхЗ и Dlx5, обнару- BMPs составляют подгруппу в сверхсемействе TGF(3 Схематическое изображение различных отделов мембранозного Внутренние пространства, заполненные эндолимфой, представлены чёрным цветом, а заполненные перилимфой белым. Серым цветом обозначены сенсорные участки (S) улитки, полукружных каналов и эллиптического и сферического мешочков.
фатическое пространство влияет также ген Gata2 [28].
живают нарушения эпителия полукружных каналов.
У наиболее тяжело пораженных индивидов формирова-
ние каналов почти полностью блокируется [11], тогда
как развитие улитки не затрагивается. У мутантов НтхЗ
и Dlx5 выпячивания, карманы для полукружных кана-
лов, формируются, но резорбция их серединных частей
неполная или задержана [47, 62]. Нарушение резорбции
может быть обусловлено изменениями в экспрессии ге-
на ВМР4 (bone morphogenetic protein 4). Возможно, что
ВМР4 регулирует клеточную гибель в этом регионе.
(Transforming growth factor beta). Предполагается, что
BMP4, -5 и -7 экспрессируются на ранних стадиях раз-
вития, участвуют в процессах формирования паттерна,
ведущего к морфогенезу полукружных каналов, а ВМР2,
по-видимому, отвечает за продолжающийся рост полу-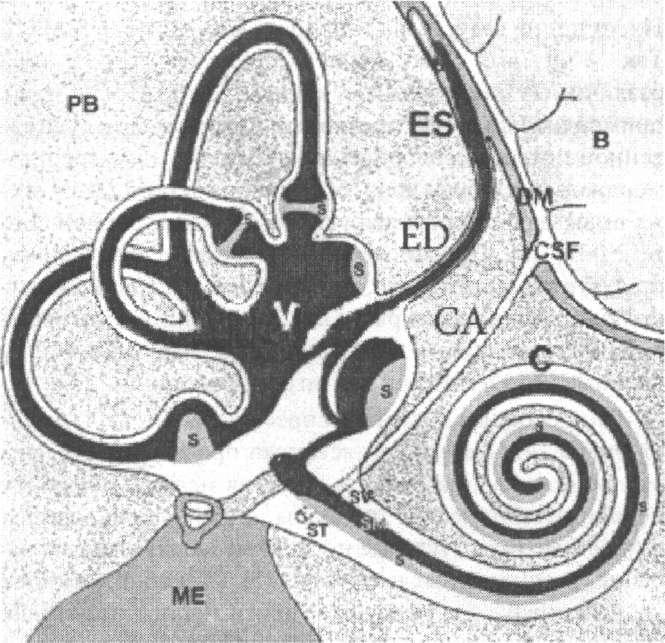
лабиринта.
В - головной мозг; С - улитка; СА - водопровод улитки (обычно не
столь широк, как показано здесь); обеспечивает общение между
спинномозговой жидкостью и перилимфатическим пространством
улитки; CSF — спинномозговая жидкость; DM — твёрдая оболочка
мозга; ED -эндолимфатический проток; ES — эндолимфатический
мешок; ME — среднее ухо; РВ — каменистая кость; S — сенсорный
орган; SM — scala media; ST - scala tympani; SV — scala vestibuli; V - вестибулярная система; эндолимфатическое пространство улитки
сообщается с таковым сферического мешочка, от которого, как и
от эллиптического мешочка, отходящие короткие протоки соединя-
ются в эндолимфатический канал (ED), который заканчивается ES
под твердой оболочкой мозга (DM).
кружных каналов после их возникновения [13]. Антаго- Нокаутные по пяти разным генам мыши, Nkx5.i Мутации, затрагивающие Иногда морфология полукружных каналов и мешоч- Два мутантных Sox2 (SRY (sex determining regi- 4
нист BMP, noggin, строго ингибировал образование по-
лукружных каналов. Он даже блокировал выпячивание
определённого индивидуального капала в зависимости
от расположения источника noggin [25].
(NK homeobox 5.1) или НтхЗ (Homeobox protein Nkx-5. I),
netrin, Otx-I, Dlx5 и Nor-1 (Neuron-derived orphan recep-
tor /), так же как и многие другие мутанты по этим ге-
нам, демонстрируют их потенциальную роль в морфоге-
незе полукружных каналов [5, 27, 47, 48, 54, 62]. Локаль-
но пролиферирующая мезенхима давит на противопо-
ложные стенки каждого из канальных выпячиваний, по-
ка стенки не сомкнутся. Netrin-1, по-видимому, являет-
ся критическим фактором в процессе соединения сре-
динных частей канальных выпячиваний, тогда как
Nkx5-1 может регулировать размер домена экспрессии
netrin-1. Резорбция середины канального выпячивания
контролируется, по-видимому, также генами Nkx5-I и
Dlx5. Их взаимодействие вместе с передачей сигналов
BMP, возможно, предопределяет область клеточной ги-
бели или рекрутирования клеток этой области в эпите-
лий формирующихся каналов. Дальнейший рост сфор-
мированных полукружных каналов регулируется орфа-
новым ядерным рецептором Nor-1 [54]. Дефицитные
мыши Gbx2(Gastrulation brain homeobox2) обнаруживают
нормальную индукцию внутреннего уха, однако позднее
в развитии ВА у них обнаруживаются аномалии [78].
спецификацию сенсорных участков
ков изменена в результате нарушения спецификации и
дифференцировки в них сенсорных участков, гребеш-
ков в ампулах полукружных каналов или макул сенсор-
ных мешочков, как это отмечалось выше у мутантов
Eyal. Нулевые мутантные мыши Foxgl (Forkheadbox GI)
обнаруживают отсутствие сенсорного гребешка и воло-
сковых клеток горизонтального полукружного канала
[23]. Известно участие сигналов Notch в определении
ранних границ сенсорных участков. Это подтверждается
наблюдениями мутантных по локусу лиганда Jag J (Jag-
ged 1) для рецептора Notch у мышей: headturner [36] и
slalom [73]. У таких мышей некоторые гребешки вести-
булярной системы маленькие или отсутствуют, паттерн
волосковых клеток в органе Корти также аномален.
У этих мутантов наблюдается последующее укорочение
заднего и переднего полукружных каналов, связанное с
отсутствием или сильным уменьшением гребней и окру-
жающих их ампул. Оба эти фенотипа могут возникать в
результате дефектов дифференциальной адгезии во вре-
мя инициальной спецификации сенсорных регионов.
on Y)-box 2) аллеля, Lcc (light coat and circling) и Ysb (yel-
low submarine) вызывают тяжёлые нарушения слуха (Ysb