Нейросенсорная глухота. 2. Генетические нарушения стероцилий
волосковых клеток
Учреждение Российской академии медицинских наук Медико-генетический научный центр РАМН,
Россия, 115478, Москва, ул., Москворечье, д.1. E-mail: mglinetz@med-gen.ru
Анализируются структура и функция стереоцилий и генетическая обусловленность разных форм
синдромальной и несиндро-мальной потери слуха у человека и животных, связанных с нарушениями
компонентов стереоцилий волосковых клеток кортиева органа. Структура и функция стереоцилий
контролируются многочисленными генами. Выявляется сеть генов, регулирующих образование,
поддержанием функционирование стереоцилий волосковых клеток, мутации
которых приводят к нарушениям слуха.
Ключевые слова: нейросенсорная глухота, стереоцилии, волосковые клетки, гены
|
Введение |
|
|
НАУЧНЫЕ ОБЗОРЫ
повторы для связывания мономерного актина, SH3-, АТФ- и Р1Р2-домены, указывая тем самым, что белок играет активную роль в сборке актина [132, 176]. Одни изоформы эспина включаются в кончик паракристалли-ческого актинового стержня и смещаются к основанию с той же скоростью, что и актиновые мономеры, другие изоформы отвечают за удлинение пучков актина [129]. |
|
|
МЕДИЦИНСКАЯ ГЕНЕТИКА. 2012. №12
хронизированную реакцию каналов механотрансдукции в пучке стереоцилий [52, 64|. Направленный на минус-концы миозин 6 и белок тирозин фосфатазного рецептора Q (PTPRQ) располагаются в основании стерео-цилии и регулируют образование и поддержание такого сужения. Миозин 6 необходим для удержания PTPRQ и, возможно, других связывающих актин белков, таких, как радиксин [104], в области сужения, где они могут соединять мембрану стерсоцилии с цитоскелетом волосковой клетки. PTPRQ может также регулировать ремоделирова-ние в этом месте актина. Он содержит фосфатидилино-зитол фосфатазный (PIPase) и тирозин фосфатазный домены. Активность PIPase домена PTPRQ обеспечивает
|
|
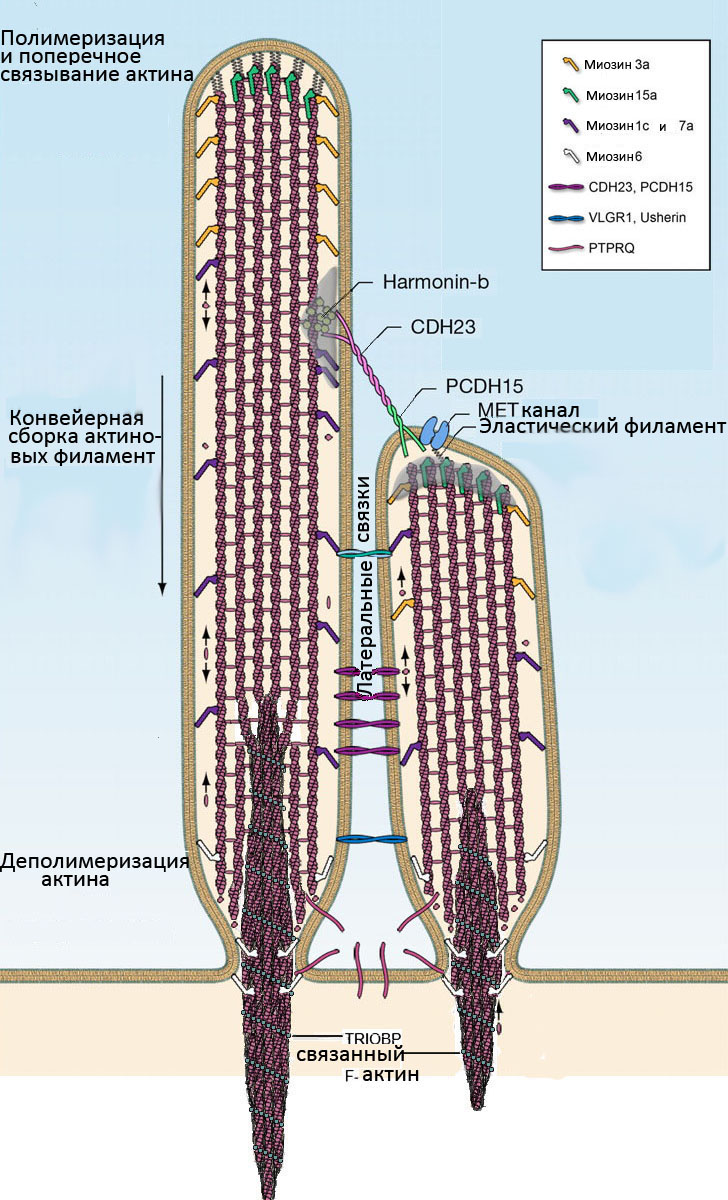
|
НАУЧНЫЕ ОБЗОРЫ
ответствующий пул актиновых мономеров для поддержания постоянной скорости обновления F-актина.
|
|
|
МЕДИЦИНСКАЯ ГЕНЕТИКА. 2012. №12
адаптация зависит от адаптационного двигательного белка, который, как полагают, состоит из кластера мио-зиновых белков, прикреплённых к верхнему концу кон-чиковой связки. Предполагается, что во время активации натяжение запорной пружины увеличивается и передаётся к двигательному белку Са2+-зависимым образом и натяжение нормализуется [41, 53, 120, 121]. |
|
|
НАУЧНЫЕ ОБЗОРЫ
клеток. Пока известен только один член семейства форминов, mDial, который участвует в возникновении не-синдромальной прогрессирующей потери слуха DFNA1 [80]. Этот формин — человеческий гомолог гена diaphanous дрозофилы, который регулирует полимеризацию актина, тем самым участвуя в поддержании актинового цитоскелета волосковых клеток [178]. Считается, что cofilin выступает в качестве важного регулятора оборота актиновых филамент. |
|
|
МЕДИЦИНСКАЯ ГЕНЕТИКА. 2012. №12
рей слуха у людей USHl: USHIB (MYOVUA), USII 1C (Harmonin), USHID (CDH23), USHIF (PCDH15) и USHIG (sans) [65, 150). Роль этих генов в формировании и функции стереоцилий описана выше. |
|
|
НАУЧНЫЕ ОБЗОРЫ
танцую рецессивную мутацию, вызывающую глухоту, поведение кружения, трясение головой и гиперактивность. В улитке наблюдается также вторичная дегенерация нейронов спирального ганглия. У других спонтанных Pcdh 15-мутантов, возникших в результате инсер-ции остатка цитозина, которая приводила к сдвигу рамки считывания и образованию преждевременного стоп-кодона, фенотип был очень схожим, хотя мыши были тугоухими [48, 159]. |
|
|
МЕДИЦИНСКАЯ ГЕНЕТИКА. 2012. №12
7А обусловливают глухоту DFNB2 у пациентов с синдромом Ашера типа IB и у мышей shakerl (Муо7а) [39, 135, 160]. В то время как мыши, гомозиготные по нулевым мутациям Муо7а, глухи, гетерозиготы имеют нормальный фенотип. Правда, миссенс-мутация гена Муо7а у мышей, трясущих головой (Hdb), вызывает фенотипические отклонения вестибулярного аппарата и умеренную потерю слуха также у гетерозигот, в результате удлинения и слияния стереоцилий волосковых клеток. Гомозиготы по этой мутации имеют более тяжёлый фенотип [122]. |
|
|
НАУЧНЫЕ ОБЗОРЫ
Гены с неизвестной функцией
Мутации гена стереоцилина STRC вызывают нарушения в локусе DFNB16, сцепленного с хромосомой 15ql5-q21, отвечающего за несиндромальную, аутосом-но-рецессивную глухоту. Ген STRC экспрессируется почти исключительно во внутреннем ухе и совпадает с некоторыми геномными клонами из хромосомной области человека DFNB16. Показано, что локус DFNB16 может содержать второй ген глухоты. Ген STRC содержит 29 кодирующих экзонов и, как было установлено, является тап-демно удвоенным со стоп-кодоном в 20-м экзоне в копии В, которая может быть псевдогеном. Предполагаемый белок стереоцилин не обнаруживает существенной гомологии с другими известными белками. Иммунофлюоресцен-тные исследования показали, что во внутреннем ухе мышей стереоцилин экспрессируется только в сенсорных во-лосковых клетках с интенсивным окрашиванием пучков стереоцилий [107]. Функция белка пока неизвестна. |
|
|
МЕДИЦИНСКАЯ ГЕНЕТИКА. 2012. №12 < hr />
Список литературы |
|
|
НАУЧНЫЕ ОБЗОРЫ
38. Gagnon L.H., Lohgo-Guess С.М., Berryman M. et al. The Chloride Intracellular Channel Protein CLIC5 Is Expressed at High Levels in Hair Cell Stereocilia and Is Essential for Normal Inner Ear Function // The Journal of Neuroscience. — 2006. — Vol. 26(40). — P. 10188-10198. |
|
|
МЕДИЦИНСКАЯ ГЕНЕТИКА. 2012. №12
75. Liu X.Z., Ouyang Х.М., Xia X.J. et al. Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss // Hum. Mol. Genet. - 2003. - Vol. 12. - P. 1155-1162. |
|
|
НАУЧНЫЕ ОБЗОРЫ
112. Prosser Н.М., Rzadzinska А.К., Steel К.P., Bradley А Mosaic complementation demonstrates a regulatory role for myosin Vila in actin dynamics of stereocilia // Mol. Cell. Biol. — 2008. — Vol. 28. - P. 1702-1712. |
|
|
МЕДИЦИНСКАЯ ГЕНЕТИКА. 2012. №12
151. van Wijk Е., Krieger Е., Kemperman М.Н. е( al. A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26) // J. Med. Genet. — 2003. — Vol. 40(12). - P. 879-884. |
|
2. Genetic disorders stereocily hair cells
Medical Genetics Research Center,
Russia, 115478, Moscow, st. Moskvorechye, 1. E-mail: mglinetz@med-gen.ru
The structure and function of stereocilia and a genetically determined different forms of syndromal and nonsyndromal
loss of hearing in humans and animal models that involve violations of the components of the stereocilia of hair cells of Corti
organ. Structure and function of stereocilia is controlled by numerous genes. Revealed a network of genes
regulating the formation, maintenance of the stereocilia of hair cells, mutations of which
lead to hearing damage.
Key words: sensorineural deafness, stereocily, hair cells, genes