James R. Lupski
Trends in Genetics 1998, 14:No. 10.417-422
[2] Higgs D.R. et al. (1980)
Nature, 284:632-635.
[3] Lauer J., Shen C.K.J. and Maniatis T. (1980)
Cell, 20:119-130.
[4] Weatherall, D.J., Clegg, J.B., Higgs, D.R. and Wood, W.G. (1995) in The Metabolic and Molecular Bases of Inherited Disease (7th edn) (Scriver, C.R., Beaudet, A.L., Sly, W.S. and Valle, D., eds), pp. 3417-3484, McGraw-Hill
[5] Metzenberg A.B., Wurzer G., Huisman T.H.J. and Smithies O. (1991)
Genetics, 128:143-161.
[6] Rayssiguier C., Thaler D.S. and Radman M. (1989)
Nature, 342:396-401.
[7] Vnencak-Jones C.L., Phillips J.A., Chen E.Y. and Seeburg P.H. (1988)
Proc. Natl. Acad. Sci. U. S. A., 85:5615-5619.
[8] Vnencak-Jones C.L. and Phillips J.A. (1990)
Science, 250:1745-1748.
[9] Steen V.M., Molven A., Aarskog N.K. and Gulbrandsen A.K. (1995)
Hum. Mol. Genet., 4:2251-2257.
[10] Nathans J. et al. (1986)
Science, 232:203-210.
[11] Motulsky, A.G. and Deeb, S.S. (1995) in The Metabolic and Molecular Bases of Inherited Disease (Vol. II) (Scriver, C.R., Beaudet, A.L., Sly, W.S. and Valle, D., eds), pp. 4275-4295, McGraw-Hill
[12] Lifton R.P. et al. (1992)
Nature, 355:262-265.
[13] Donohoue P.A., Jospe N., Migeon C.J. and Van Dop C. (1989)
Genomics, 5:397-406.
[14] Simon D.B. et al. (1997)
Nat. Genet., 17:171-178.
[15] Zimran A. et al. (1990)
J. Clin. Invest., 85:219-222.
[16] Yen P.H. et al. (1990)
Cell, 61:603-610.
[17] Ballabio A. et al. (1990)
Genomics, 8:263-270.
[18] Li X-M., Yen P.H. and Shapiro L. (1992)
Nucleic Acids Res., 20:1117-1122.
[19] Lupski J.R. (1997)
Hosp. Pract., 32:83-122.
[20] Lupski J.R. (1998)
Mol. Med., 4:3-11.
[21] Lupski, J.R. (1998) in Scientific American Molecular Neurology (Martin, J.B., ed.), pp. 239-256, Scientific American Inc.
[22] Pentao L. et al. (1992)
Nat. Genet., 2:292-300.
[23] Chance P.F. et al. (1994)
Hum. Mol. Genet., 3:223-228.
[24] Reiter L.T. et al. (1997)
Hum. Mol. Genet., 6:1595-1603.
[25] Kiyosawa H. and Chance P. (1996)
Hum. Mol. Genet., 5:745-753.
[26] Kiyosawa H., Lensch M.W. and Chance P.F. (1995)
Hum. Mol. Genet., 4:2327-2334.
[27] Timmerman V. et al. (1997)
J. Med. Genet., 34:43-49.
[28] Lopes J. et al. (1996)
Am. J. Hum. Genet., 58:1223-1230.
[29] Reiter L.T. et al. (1996)
Nat. Genet., 12:288-297.
[30] Reiter L.T. et al. (1998)
Am. J. Hum. Genet., 62:1023-1033.
[31] Lopes J. et al. (1998)
Hum. Mol. Genet., 7:141-148.
[32] Palau F. (1993)
Hum. Mol. Genet., 2:2031-2035.
[33] Lopes J. et al. (1997)
Nat. Genet., 17:136-137.
[34] Chen K-S., Potocki L. and Lupski J.R. (1996)
Mental Retard. Dev. Disab. Res. Rev., 2:122-129.
[35] Greenberg F. et al. (1996)
Am. J. Med. Genet., 62:247-254.
[36] Greenberg F. et al. (1991)
Am. J. Hum. Genet., 49:1207-1218.
[37] Guzzetta V. et al. (1992)
Genomics, 13:551-559.
[38] Juyal R.C. et al. (1996)
Am. J. Hum. Genet., 58:998-1007.
[39] Chen K-S. et al. (1997)
Nat. Genet., 17:154-163. ]
[40] Dutly F. and Schinzel A. (1996)
Hum. Mol. Genet., 5:1893-1898.
[41] Urban Z. et al. (1996)
Am. J. Hum. Genet., 59:958-962.
[42] Perez-Jurado L.A. et al. (1996)
Am. J. Hum. Genet., 59:781-792.
[43] Robinson W.P. et al. (1996)
Genomics, 34:17-23.
[44] Wu Y. et al. (1998)
Am. J. Med. Genet., 78:82-89.
[45] Osborne L.R. et al. (1997)
Genomics, 45:400-404.
[46] Carrozzo R. et al. (1997)
Am. J. Hum. Genet., 61:228-231.
[47] Christian S.L. et al. (1995)
Am. J. Hum. Genet., 57:40-48.
[48] Christian S.L. et al. (1997)
Am. J. Hum. Genet., 61:A24.
[49] Morrow B. et al. (1995)
Am. J. Hum. Genet., 56:1391-1403.
[50] Lindsay E.A. et al. (1995)
Am. J. Med. Genet., 56:191-197.
[51] Halford S. et al. (1993)
Hum. Mol. Genet., 2:191-196.
[52] Morrow B.E. et al. (1997)
Am. J. Hum. Genet., 61:A25. ]
[53] Perez-Jurado L.A. et al. (1998)
Hum. Mol. Genet., 7:325-334.
[54] Lakich D., Kazazian J.H.H. Jnr, Antonarakis S.E. and Gitschier J. (1993)
Nat. Genet., 5:236-241.
[55] Naylor J.A. et al. (1995)
Hum. Mol. Genet., 4:1217-1224.
[56] Rossiter J.P. et al. (1994)
Hum. Mol. Genet., 3:1035-1039.
[57] Bondeson M-L. et al. (1995)
Hum. Mol. Genet., 4:615-621.
[58] Timms K.M. et al. (1997)
Hum. Mol. Genet., 6:479-486.
[59] Lagerstedt K. et al. (1997)
Hum. Mol. Genet., 6:627-633.
[60] Rudiger N.S. et al. (1991)
Clin. Genet., 39:451-462.
[61] Olds R.J. et al. (1993)
Biochemistry, 32:4216-4224.
[62] Kornreich R., Bishop D.F. and Desnick R.J. (1990)
J. Biol. Chem., 265:9319-9326.
[63] Marcus S. et al. (1993)
Hum. Genet., 90:477-482.
[64] Pousi B. et al. (1994)
Am. J. Hum. Genet., 55:899-906.
[65] Shen P. and Huang H.V. (1986)
Genetics, 112:441-457.
[66] Vulic M., Dionisio F., Taddei F. and Radman M. (1997)
Proc. Natl. Acad. Sci. U. S. A., 94:9763-9767.
[67] Donohoue, P.A., Parker, K. and Migeon, C.J. (1995) in The Metabolic and Molecular Bases of Inherited Disease (Vol. II) (Scriver, C.R., Beaudet, A.L. and Valle, D., eds), pp. 2929-2966, McGraw-Hill
[68] Wise C.A. et al. (1993)
Am. J. Hum. Genet., 53:853-863.
[69] Nelis E. et al. (1996)
Eur. J. Hum. Genet., 4:25-33.
[70] Lupski J.R., Roth J.R. and Weinstock G.M. (1996)
Am. J. Hum. Genet., 58:21-27.
[71] Melki J. et al. (1994)
Science, 264:1474-1477.
[72] Lefebvre S. et al. (1995)
Cell, 80:155-165.
[73] Wang C.H. et al. (1995)
Am. J. Hum. Genet., 56:202-209.
[74] Konrad M. et al. (1996)
Hum. Mol. Genet., 5:367-371.
[75] Inoue K. et al. (1996)
Am. J. Hum. Genet., 59:32-39.
[76] Lagerström-Fermer M. et al. (1997)
Am. J. Hum. Genet., 60:910-916.
[77] Small K., Iber J. and Warren S.T. (1997)
Nat. Genet., 16:96-99.
[78] Small K. and Warren S.T. (1998)
Hum. Mol. Genet., 7:135-139.
[79] Wijmenga C. et al. (1992)
Nat. Genet., 2:26-30.
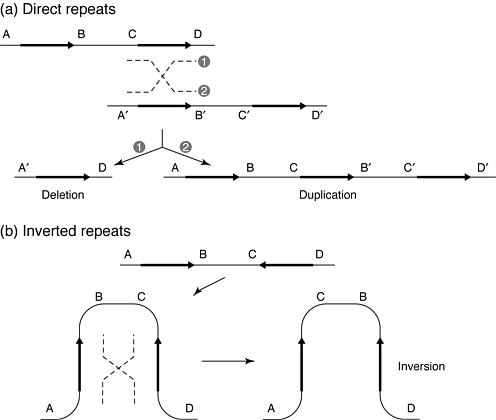 |
Рис. 1. Геномные перестройки в результате рекомбинации между повторяющимися последовательностями. Направление повторов показано стрелками. Большими буквами выше и ниже тонкой горизонтальной линии обозначены уникальные фланкирующие последовательности. Щтриховые линии означают событие рекомбинации, результаты которой представлены цифрами 1 и 2. (a) Рекомбинация между прямыми повторами дает делецию и/или дупликацию. (b) Рекомбинация между инвертированными повторами дает инверсию. Figure 1. Genomic rearrangements resulting from recombination between repeated sequences. Repeated sequences are depicted as black arrows with the orientation indicated by the direction of the arrowhead. Capital letters above or below the thin horizontal lines refer to the flanking unique sequence (e.g. A). Chromosome homologs are also shown (e.g. A9). Dashed lines refer to a recombination event with the results shown by numbers 1 and 2. (a) Recombination between direct repeats results in deletion and/or duplication. (b) Recombination between inverted repeats results in an inversion. |
Types of genomic disorders
Tandemly repeated genes
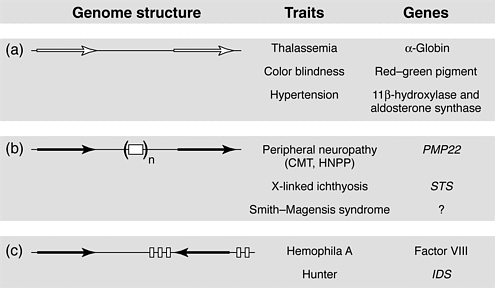 |
Рис. 2. Структурные свойства генома и примеры геномных нарушений. (a) Тандемно повторенные гены. Свойства генома (геномная структура) предсталвены генами, указанными открытыми стрелками. Примеры признаков заболеваний, которые могут быть вызваны этой геномной архитектурой представлены генами, затрагимваемыми перестройками. (b) Тандемные перестройки отдельные от генов. Гены с дозовой чувствительностью (открытые горизонтальные четырехугольники) или гены (n>1) фланкированные повторами(черные стрелки) в тандемной ориентации. Если рекомбинация происходит между неправильно расположенными фланкирующими повторами (неравный кроссинговер) то гены между повторами могут быть делетированы или удвоены. Примеры заболеваний, которые появляются при такой геномной архитектуре и являющиеся результатом генной гаплонедостаточности (делеции) или увеличения дозы гена (дупликации), даны с определенными вовлеченными генамиare [? референтные чувствительные к дозе гены неизвестны]. (c) Инвертированные повторы с одной копией повтора внутри гена. Много-экзонные гены с одной копией повтора (черная стрелка) между экзонами (белые четырехугольники) а дополнительная клопия повтора расположена или выше или ниже гена. Эта геномная архитектура вызывает инверсию, которая нарушает интегральность структуры . Figure 2. Genome structural features and example genomic disorders. (a) Tandemly repeated genes. The features of the genome are shown (genome structure) with genes indicated as open arrows. Examples of disease traits that might be caused by this genomic architecture are given with the genes affected by the rearrangement. (b) Tandem repeats separated from genes. A dosage-sensitive gene (open horizontal rectangle) or genes (n>1) is flanked by a repeat (black arrows) in tandem orientation. If recombination occurs between mis-aligned flanking repeats (unequal crossing over) the genes located between the repeats can be deleted or duplicated. Examples of diseases that can occur because of this genomic architecture, and be the result of gene haploinsufficiency (deletion) or increased gene dosage (duplication) effects, are given with the particular gene involved [? Refers to dosage-sensitive gene(s) unknown]. (c) Inverted repeats with one copy of the repeat located within a gene. A multi-exon gene with one copy of a repeat (black arrow) located between exons (white rectangles) but an additional copy of the repeat is located either upstream or downstream from the gene. This genomic architecture can result in an inversion that disrupts the structural integrity of the gene. |
β
Цитохром P450 фермент debrisoquine 4-hydroxylase (CYP2D6) метаболизирует многочисленные различные классы лекарств. Терапевтическая эффективность и побочные эффекты метаболизируемых лекарств с помощью CYP2D6 генного продукта может зависеть от индивидуального генотипа в этом локусе. CYP2D6, - член CYPD кластера из трех тандемно расположенных генов, фланкируемчх повтором в 2.8 kb. Точки разрывов при общераспространенном делеционном аллеле, ассоциирующем с плохим метаболизмом, обнаруживаются внутри повтора 9.
Гены кодирующие альдемтерон синтазу и steroid 11βhydroxylase на 95% идентичны и располагаются на расстоянии в 45 kb на хромосоме 8q. Glucocorticoid-remediable aldosteronism (GRA) является аутосомно доминантным нарушением, которое характся гипертензией с варьирующим гиперальдостерпонизмом и высоким уровнем аномальных стероидов надпочечника, которые находятся под контролем адренокортикотропного гормона и супрессируемых глюкокортикоидами. GRA обусловлен удвоением гена, возникающим в результате неравного кроссинговера, в результате которого сливается 5' регуляторная область гена, кодирующего 11βhydroxylase (11-OHase)с кодирующими последовательностями aldosterone synthase (AldoS). В результате возникает несоответствующая экспрессия альдестерон синтазы в тканях или во время, когнда она не должна экспрессироваться 12.
Другими геномными нарушениями - результатом неравного кроссинговера физически сцепленных повторяющихся генов или гена и сходного псевдогена являются: (1) 21-hydroxylase нехватка благодаря рекомбинации между CYP21 генами13; (2) Bartter syndrome type III, следствие рекомбинации между родственными генами, кодирующими хлорные канальцы , CLCNKB и CLCNKA (Ref. 14); and (3) Gaucher disease благодаря рекомбинации между геном для acid βglucosidase и соседним псевдогеном 15.
Tandem repeats separated from genes
Один из наиболее охарактеризованных геномных нарушений с помощью рекомбинации между танденмными повторами является нейропатия Charcot-Marie-Tooth disease type 1A (CMT1A), которая ассоциирует с 1.5 Mb тандемной дупликацией в 17p12 (Refs 192021). Эта дупликация возникает в результате неравного кроссинговера и гомологичной рекомбинации между 24 kb фланкирующими повторами, называемыми CMT1A-REP (Ref. 22). Реципрокный рекомбинантный продукт с участием CMT1A-REP дает в результате делецию в 1.5 Mb , которая ассоциирует с клинически отличающейся периферической нейропатией -наследственной нейропатией с лабильностью к давлению ( liability to pressure palsies (HNPP))23. CMT1A и HNPP результат нарушения копийности гена миэлина с чувствительностью к дозеPMP22, который локализуется 0.5 Mb от проксимальной или центромерной копии CMT1A-REP и 1.0 Mb от дистальной или теломерной CMT1A-REP. Две CMT1A-REP копии имеют общие 24 011 п.н. из 98.7% последовательностей 24. Повтор CMT1A-REP , по-видимому, удвеивается во время эволюции генома приматов, так как человек и шимпанзе имеют по две копии, тогда как гориллы и другие низшие приматы имеют только одну копию 2425. Таким образом, эволюция генома млекопитающих может создавать структурные свойства, делающие определенные области генома человека чувствительными к геномным нарушениям.
Гомологичная рекомбинация между фланкирующими CMT1A-REPs ассоциирует с горячей точкой для кроссоверов,где происходит >70% всех случаев2526272829. Анализх нуклеотидных последовательностей области обмена нитями в HNPP делеции30 и в CMT1A дупликации31 индивидов выявляет, что кроссоверы появляются в длинном участке (>400 н.п.) идентичности внутри высоко гомологичных CMT1A-REPs. Длина этих участков идентичности может быть равна или выше минимального эффективно процессируемого сегмента (minimal efficient processing segments (MEPS)) для гомологичной рекомбинации в мейозе человека 30. Более того анализ рекомбинантных CMT1A-REPs полкучил доказательства событий генной конверсии, которые служат признаком двунитчатых разрывов, которые часто инициируют гомологичную рекомбинацию 30. Предполагается, что mariner-подобный элемент, называемый MITE, вместе с trans действующей транспозазой, ответственненой за инициациию двунитчатых разрывов в CMT1A локусе29. Горячие точки рекомбинации ассоциированные с дупликацией The CMT1A и делецией HNPP , по-видимому, перекрываются с самым длинным участком идентичных последовательностей между CMT1A-REPs, которые являются ближайшими к элементу MITE 2930. Другими исследователями было предположено, что другой мотив из коротких последовательностей участвует в инициации процесса рекомбинации 31. Интересно половое предпочтение для de novo CMT1A удвоений, с подавляющим больштинством удвоений в результате неравного кроссинговера во время гаметогенеза самцов 3233. Однако немногие делеции HNPP задокументированы как появившиеся во время гаметогенеза самок и возникают в результате внутрихромосомных обменов33.
Inverted repeats
Инверсия гена, кодирующего iduronate-2-sulfatase (IDS), возникающая в результате рекомбинации с IDS-родственными последовательностями является причиной синдрома Hunter 57. Псевдо ген (IDS-2) располагается на 20 kb дистальнее и в противоположной ориентации к гену IDS 5758. Локус IDS-2 тянется примерно на 3 kb и обнаруживает более чем на 88% идентичных последовательностей с IDS геном5859. Все события рекомбинации происходят внутри области в 1 kb , где идентичность последовательностей достигает более 98%59. Событие рекомбинации начинается с двойного разрыва в интроне 7 гена IDS 59.
General features regarding mechanisms for genomic disorders
- Большие области гомологии необходимы для рекомбинации. Гомологичные области обычно занимают свыше тысяч пар оснований. Короткие разбросанные последовательности, такие как Alu, обычно не являются субстратом, хотя рекомбинация с участием Alu может происходить с помощью нелегитимно или гомологичной рекомбинации и иногда может приводить или к делециям или дупликациям при неравном корссинговере 6061626364.
- Выявляется широкая вариабельность длин повторов удвоенных геномных сегментов (1). Однако, если геномные нарушения ранжировать по расстояниям между повторами, то длина повторов коррелирует позитивно с расстоянием между посторами (1). В целом, чем больше расстояние между повторами, тем значительнее длина повтора потенциально необходима для эффективной рекомбинации. Это может отражать тот факт, что для осуществления неравного кроссинговера и рекомбинации на большом расстоянии необходжима большая джлина идентичных последовательностей для стабилвизации рекомбинационного комплекса. Или напротив чем дальше расположены друг от друга повторы, тем скорее большие участки идентичных последовательностей смогут найти дриуг друга и спариться.
- Обмех хроматид или кроссинговер происходит преимущественно в области точной идентичности внутри повторов. Необходимость в значительных участках идентичных последовательностей указывает на то, что mismatch-repair и recombination machinery распознают гетерогенные последовательности внутри сходных областей и прерывают рекомбинацию натыкаясь на отсуствие идентичных последовательностей. Или возможно, что a RecA-подобный белок может быть необходим и следить за идентичностью внутри сходных областей. Эти идентичные области могут отражать MEPS , необходимую для гомологичной рекомбинации 6566.
- Двухнитчатые разрывы могут быть инициирующим событием реекомбинации между повторами, ведущей к геномным нарушениям. Анализ соединившихся последовательностей ДНК при HNPP, CMT1A и Hunter синдроме выявляет мозаичные (interspersed)участки последовательностей ДНК из двух рекомбинирующих посторов, предполагающих события генной конверсии. Это может быть результатом репарации гетеродуплексной ДНК во время разрешения Holliday junctions.
- В некоторых случаях de novo перестройки, ведущие к геномным нарушениям, обнаруживают parent-of-origin effect. Это четко задокументировано как для подавляющего большинства случаев удвоений CMT1A и случаев инверсий фактора VIII , перестройки возникают в хромосома, получаемых от отца. Эти данные указывают на то, что потребности в гомологичной рекомбинации, дающей геномные нарушения, отличны при гаметогенезе у самцов и самок.
- Не все копии данного повтора используются эксивалентно в качестве субстрата для гомологичной рекомбинации. Как показано для делеций SMS и инверсий фактора VIII , соседние посторы не обязательно вовлекаются с наибольшей частотой в рекомбинацию. Более того добавочные низкокопийные повторы идентифицированны в области SMS хромосомы 17p11.2, но фланкирующие SMS-REPs повторы, по-видимому, более предпочтительны для рекомбинации. Следовательно, и другие факторы, структурные свойства высшего порядка хромосом в синаптонемальных комплексах во время мейоза, необходимы для выравнивания гомологичных субстратов. Близость к местам инициации рекомбинации может быть также важной.
Table 1. Физические свойства областей, связанных с геномными нарушениями | ||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||
Other genomic disorders
В случаях геномных перестроек, обусловленных делециями, частоты реципрокных дупликаций могут быть недосчитаны. Если другие contiguous-gene-deletion синдромы, такие как Williams, Prader-Willi и Angelman, и DiGeorge/velo-cardio-facial синдромы, то результаты молекулярных исследований сходны с теми, что при SMS, когда могут появиться реципрокные дупликации, характерные для SMS (Ref. 39). Такие пациенты с дупликациями могут иметь другие клинические проявления и менее выраженный фенотип, чем те что с делециями, так как избыток генетической информации обычно менее вреден для организма, чем делеция. Следовательно, эти случаи будут реже идентифицироваться или могут пропускаться при рутинном цитогенатическом анализе. Хромосомные удвоения более частые мутационные изменения, чем обнаруживаются70.
Другие болезни, обычно вызываемые перестройками ДНК могут отражать уникальные структурные свойства генома. Они могут включать: спинальные мышечные атрофии, ассоциированные с повторяющимися последовательностями в 5q13 (Refs 717273); juvenile nephronophthisis (recessive medullary cystic kidney disease), ассоциированный с большими гомозиготными делециями в 2q13, с участием 100 kb инвертированной дупликации 74; удвоенный ген PLP обусловливает болезнь Pelizaeus-Merzbacher 75; региональная дупликация в Xq25-26 ассоциированная с Х-сцепленным рецессивным panhypopituitarism76; Инверсии вокруг локуса emerin, ассоциированные с 11.3 kb инвертированным повтором Xq28 (Refs 7778); и уникальное расположение последовательностей в локусе 4q35 , ассоциированном с fascioscapulohumeral muscular дистрофией79. По мере того Как дополнительные большие ДНК коследовательности contigs становятся доступными благодаря успехам проекта генома человека, сложность архитектуры генома человека будет выясняться и дальше.