Human organomics: a fresh approach to understanding human development using single-cell transcriptomics
 Developmen t (2017) 144, 1584-1587 doi:1 0.1242/dev .150458 | ||
|---|---|---|
|
|
Понимание того, как множественные отличающиеся типы клеток соединяются вместе, чтобы строить органы давно интересует биологов. Многие годы мы изучали молекулярные события, инструктирующие клеточные клоны, необходимые специфические факторы роста, и морфологические аспекты, управляющие развитием органов. Большая часть знаний была получена в исследованиях органогенеза не человеческих позвоночных; однако, наблюдение, что существуют различия между тем, как формируются органы у ряда видов поставило вопрос, что делает нас уникальными людьми. Открытие, что плюрипотентные стволовые клетки человека могут самоорганизовываться в трехмерные структуры, которые содержат множественные дифференцированные типы клеток, организованные в нечто, напоминающее первичную человеческую ткань, воскресило область биологии развития человека (McCaul ey and Wells, 2017). В целом эти структуры обозначаются как органоиды и были разработаны протоколы по генерации зачатков кишечника, почек печени, множественных регионов головного мозга человека и др. тканей (Mc Cauley and Wells, 2017). Обычные стратегии анализа развития органоидов человека часто оценивают клеточный состав и дифференцировку с помощью иммунохимии ограниченного набора маркерных белков или путем отслеживания клеток с помощью репортерных генов. Поскольку органоиды являются по определению состоящими из многих разных типов клеток и часто обнаруживают значительную вариабельность от органоида к органоиду, то высокопроизводительная транскриптомика одиночных клеток предоставляет удивительную стратегию по оценке клеточного состава, клональных взаимоотношений и генетических сетей в органоидах. Здесь мы обсудим, как органомика (organomics) человека, под которой мы понимает способ использования функциональной геномики для изучения развития органов человека на уровне одиночной клетки, может быть использована, чтобы органоиды могли улучшить наше понимание развития человека. Мы сконцентрируемся на секвенировании РНК одиночных клеток (scRNA-seq), суммируя некоторые обычно используемые методы, их преимущества и ограничения. Словарь технических терминов и акронимов см. в Box 1. Single-cell transcriptomics: one technology, many methods. Capturing RNA from single cells Начиная с плотных тканей, генеральная стратегия каждого scRNA-seq подхода начинается с диссоциации ткани на суспензию из одиночных клеток, затем с отлавливания индивидуальных клеток в изолированные компартменты, лизиса клеток, приготовления амплифицированной кДНК с РНК (обычно мРНК) и, наконец, генерации библиотеки составных последовательностей, при этом все молекулы кДНК от индивидуальной клетки, содержащие одну и ту же уникальную последовательность, наз. клеточным штрих-кодом (barcode). Существующие методы транскриптомики одиночных клеток отличаются по способу отлавливания и компартментализации, способу амплификации кДНК, по способу внесения клеточных штриховых кодов молекул в кДНК. Суммируем наиболее распространенные методы выделения клеток для приготовления кДНК и наделения клеток штриховым кодом. Для детального рассмотрения методов обращайтесь к недавним обзорам (Kumar et al., 2017; Kolodzi ejczyk et al., 2015).
Существуют множественные платформы для выделения клеток, включая valve-based microfluidics (Fluidigm C1 или home-built chips), droplet-based microfluidics [Drop-seq, inDrop, commercial brands Chromium (10x Genomics) и BDTM Resolve System (BD Genomics)], сортирующие одиночные клетки в колодцы (wells) multi-well пластинки (Sm art-seq1/2, MARS-s eq, CEL-seq 2, SCR B-seq) или случайно диспергированные клетки в колодцы microwell пластинок (комерческая марка WaferGen), каждая платформа имеет свои собственные преимущества и недостатки. Как только индивидуальные клетки оказываются изолированными, то кДНК обычно генерируется с помощью обратной транскрипции, начиная с poly-A хвостовой мРНК. Возникающая в результате кДНК затем амплифицируется или экспоненциально с помощью PCR реакции (Drop-seq, Sma rt-seq1/2, STRTseq, SCRB-seq) или quasi-linearly посредством транскрипции in vitro (CEL-se q1/2, inDrop). Базирующийся на PCR метод Smart-seq2 основывается на обратных транскриптазах с переключением матриц, которые гарантируют генерацию кДНК полной длины и секвенирование всего транскрипта (Picelli et al., 2013). Большинство др. методов обеспечивают секвенирование только 3 ' или 5 ' концевых транскриптов, которые пригодны для прикрепления уникальных молекулярных идентификаторов, способствующих подсчету индивидуальных молекул (ST RT-seq, Drop-seq, inDrop, SCR B-seq, CEL-seq, MARS-s eq) скорее, чем подсчету количества мРНК с помощью нормализации по картированному считыванию (against mapped reads). Во всех случаях основной проблемой в отлавливании клеток является генерация суспензии, так, чтобы клетки оказывались в жизнеспособными, будучи одиночными (singlets).
Многочисленные вариации в энзимах, времени диссоциации, методов титрации и т. д. были проверены с целью оптимизации ступени диссоциации. Дополнительные соображения д. учитывать возможное обогащение или исключение определенных типов клеток в некоторых протоколах диссоциации, а также тот факт, что сама природа протокола может изменять транскриптом клетки. В головном мозге, напр., дифференциальная ломкость определенных типов клеток после диссоциации ткани может означать, что сбор и профилирование может обнаруживать смещение в направлении более крепких клеток, таких как астроциты и нейральные предшественники. Это также составляет проблему при использовании scRNA-seq для определения качественных особенностей представленных клеток, поскольку ломкие клетки могут экспрессировать маркеры гибели клеток или клетки могут становиться активированными во время разрушения ткани. Наконец, некоторые типы клеток, напр., панкреатические клетки, могут содержать энзимы, влияющие на синтез кДНК лизированными одиночными клетками. Методы секвенирования транскриптома ядер отдельных клеток составляют привлекательную стратегию для борьбы с проблемой ломкости и пертурбаций экспрессии генов во время стадии отлавливания РНК, поскольку ядра являются довольно крепкими и могут быть легко очищены с помощью FACS (Habib et al., 2016).
Среди всех этих доступных методов биологи и д. выбирать для изучения формирования органоидов на транскрипционном уровне? Три ключевые переменные следует учитывать: количества собранных клеток, глубину секвенирования (т.е., количество считываний или транскриптов на клетку) и величину покрытия транскриптома. Чтобы анализировать аллель-специфичную экспрессию или альтернативный сплайсинг, необходимо выбрать метод, обеспечивающий покрытие всего генома, напр., Smart-seq2. Такое высокое покрытие всего транскриптома дорого, однако, поскольку многие секвенируемые транскрипты (reads) необходимы для одного и того же гена, то это ограничивает производительность клеток. Высокая производительность клеток является критической в отношении реконструкции развития органоидов и сравнима среди органоидов, индуцируемых с помощью линий плюрипотентных стволовых клеток, условий или индивидов. В перспективе секвенирования, клеточная производительность может быть усилена с помощью секвенирования только концов транскриптов (Jaitin et al., 2014). Это будет конечно зависеть от типов клеток и степени гетерогенности внутри органоидов. При сортировке пластинок ручная работа может быть уменьшена при использовании жидкостной робототехники. Капельно-микроструйные (droplet- microfluidics) подходы предоставляют существенное улучшение перед предыдущими технологиями, драматически увеличивая производительность клеток путем снижения стоимости ручной работы ( Klein et al., 2015; Macosko et al., 2015). Имеются и др. высокопроизводительные подходы на горизонте, базирующиеся на капельных методах (Rosenber g et al., 2017 preprint; Cao et al., 2017 preprint). Необходимо отметить, что высокопроизводительные подходы часто обнаруживают меньше генов на клетку, чем высоко-покрывающие подходы и могут, следовательно, не выявлять всей гетерогенности, которая определяется лишь немногими генами или генами, экспрессируемыми на низких уровнях. Чтобы обойти это ограничение, возможно необходимо объединить экспрессию генов всех клеток в кластер и генерировать средние транскриптомы, чтобы достичь чувствительности высоко-покрывающих технологий. Defining the cellular composition of organoids Сила транскриптомики одиночных клеток базируется на её способности идентифицировать молекулярно отличающиеся типы клеток в комплексе ткани без предварительной очистки типов клеток. Это зависит в наибольшей степени от математических подходов, используемых для идентификации разных клеточных популяций. Анализ принципиальных компонентов (PCA) широко используется для идентификации генов, варьирующих в собранных одиночных клетках. Клеточные взаимоотношения затем могут быть визуализованы в двумерном пространстве на основании экспрессии этих генов, напр., с использованием tSNE или графов, указывающих направление сил,и сгруппированного использования разных отличающихся алгоритмов, таких как BackSPIN , K-means и др. ( Kumar et al., 2017; Kolodzi ejczyk et al., 2015). Главной проблемой по деконструкции клеточного состава, это понимание источника гетерогенности в базе данных. Технические шумы, главным образом, пропадание информации ('dropout ') обусловлено неэффективностью отлавливания мРНК и групповыми эффектами, могут смущать анализ и затемнять настоящие биологические шумы. Имеются стратегии для борьбы с техническими шумами и по удалению смешанных переменных, такие ка состояние клеточного цикла (Steg le et al., 2015). Кроме того, часто неясно, каковы составляющие клеточных типов и как определить дискретный тип клеток в противовес непрерывным клеточным состояниям. Как результат отсутствует окончательное решение, где проходит линия между разными клеточными состояниями. Важно многократное исследование данных с использованием множественных подходов для полного понимания источников гетерогенности в базах данных. Также в целях идентификации редких типов клеток необходимо проанализировать многие тысячи клеток или редкие типы клеток следует обогатить, используя FACS, центрифугирование в градиенте плотности или др. подходы. Эти методы диссоциации могут обогатить или исключить определенный тип клеток из суспензии; поэтому в целом неясно, действительно ли scRNA-seq может надежно оценивать количества типов клеток. Reconstructing lineage relationships with in an organoid Органоиды содержат незрелые и зрелые клетки при одном и том же возрастном уровне дифференцировки. scRNA-seq осуществляет снимок состояний транскриптома, присутствующих в ткани в любой данный момент. В недавнем исследовании коры человеческих плодов и органоидов scRNA-seq выявило присутствие промежуточных клеток, которые могли бы способствовать реконструкции пути клонирования от предшественника до нейрона (Fig. 1) ( Camp et al., 2015). Следовательно, основной силой данной технологии является выявление линий возрастных траекторий посредством присутствия промежуточных состояний. Это привело к созданию разным математических моделей, которые пытаются описать клетки в псевдовременном порядке, а экспрессия генов может быть отслежена как функция псевдовремени (pseudotime) (Trapn ell et al., 2014; Setty et al., 2016; Haghverdi et al., 2016). Кроме того, возможно также определить бифуркации клонов, когда общий предшественник дает два или более дифференцированных типа клеток. 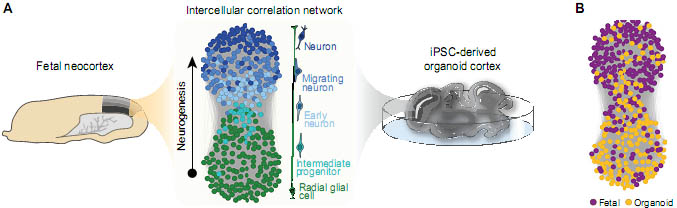 Fig. 1. Human organomics: reconstructing organoid development using scRNA-seq. (A) scRNA-seq comparison s between fetal human tissue and organoids bring insight into how well organoids recapitul ate human development. As an example, scRNA-seq was used to analyze human fetal neocort ex a nd cerebral organoids (Camp et al., 2015). An intercellular correlation network between fetal and organoid cells can reconstruct cell line age relatio nships between radial glial progenitors, intermediates, and neurons. Th e network is colored based on the cell type. (B) Fetal and organoid cells are highly correlat ed and intermix in the network. The network is colored based on whet her the cell is derived from the organoid or fetal cortex. iPSC, induced pluripotent stem cell. Challenges and opportunities in human organomics Современные методы понимания выбора клонов с использованием scR N A-seq не предоставляют прямых доказательств взаимоотношений между клонами, т.к. клоны реконструируются посредством перекрывающихся паттернов экспрессии генов в клетках на промежуточных стадиях. Существует несколько методологических ухищрений, позволяющих отслеживать непосредственно клональные взаимоотношения с использованием транскриптомики одиночных клеток в органоидах. Одна из стратегий - это использование библиотеки вирусов, чтобы инфицировать клетки, экспрессирующих в каждой клетке репортер с уникальным barcode ( Gerlach et al . , 2 0 13 ). Измерения транскриптома одиночной клетки д. включать клон-определяющий-barcode, как и транскриптом. Др. стратегия заключается в индукции целенаправленных мутаций в ДНК с использованием CRISPR/Cas9 для создания уникальных паттернов инсерций и делеций, известных как клеточно-специфические маркеры, передающиеся дочерним клеткам ( Mc Kenna et al. , 2 0 16 ). Клональный транскриптом при своем образовании может быть считан или с помощью секвенирования ДНК и РНК из одной и той же клетки или с помощью создания мутаций в экспрессируемом гене, напр., совместно с флюоресцентным репортером, и только в секвенируемом транскриптоме ( Ju ker et a l. , 2 0 1 7 preprint) . Spatial transcri ptomics Сегодня широко используемые методы scRNA-seq нуждаются в диссоциации ткани на суспензию из одиночных клеток, чтобы компартментализовать клетки для barcoding. Однако, этот подход лишен пространственного разрешения. В недавнем обзоре было объяснено, как многие лаб. демонстрируют измерения на транскриптомной шкале количеств РНК внутри ткани (Tanay and Regev, 2 017 ; Crosetto et al. , 2 01 5) . Один из подходов для пространственного разрешения транскриптомов одиночных клеток связан с комбинацией данных scRNA-seq с reference картами, сгенерированными с помощью традиционной in situ гибридизации. Др. подход использует последовательные раунды smFISH с утонченным комбинаторным флюоресцентным barcoding, чтобы оценить количественно экспрессию десятков-сотен генов in situ ( Shah et a l. , 2 0 1 6) . Дополнительной стратегией является определение позиции гистологических срезов на праймерах обратной транскрипции с уникальными позиционными barcodes, тем самым генерируются данные по секвенированию РНК с сохранением двумерного расположения позиционной информации (Stahl et al., 2016). Эти подходы выявляют расположение определенных типов клеток и предоставляют мощные преимущества над методами, базирующимися на диссоциации. Поскольку органоиды относительно гетерогенны и могут быть лишены стереотипичной онтогенетической оси, локализация транскриптомов внутри архитектуры ткани д. быть очень полезной. Технические затруднения и необходимые экспертизы для пространственной транскриптомики затрудняют их широкое использование; , однако, это может измениться в будущем. Beyond cell atlases: mechanisms of human development and disease Главной целью транскриптомики одиночных клеток человека является генерация исчерпывающего и количественного описания ' атласа ' каждого типа клеток человека взрослых и плодных тканей. Атлас клеток человека представляет описание для сравнения с органоидами, чтобы понять, как клеточный состав и сети генов воспроизводятся в этих моделях in vitro. Кроме того, атласы клеток плодов человека может обеспечивать стратегии по обратному инженерингу тканей человека путем идентификации транскрипционных факторов, специфичных для определенных типов клеток, и предоставлению информации о межклеточных коммуникациях посредством определения предполагаемых пар рецептор-лиганд. Более того, сравнения между тканями человека при болезни и атласом сможет помочь по выяснению механизмов болезней на уровне клеток и может даже открыть 'новые ' типы клеток человека. исследования, такие как эти, описывают явления, тогда как человеческие органоиды открывают возможность выявления регуляторных механизмов путем исследования и нарушений процессов развития в контролируемых условиях. Как таковые, измерения с высоко информативным содержанием и сложные математические подходы по позиционированию scRNA-seq являются мощными инструментами по выяснению онтогенетических механизмов, которые могут быть протестированы. Сравнения иежду или органоидами, сгенерированными от здоровых и больных индивидов с использованием высокопроизводительной, пространственной и связанной с возрастом транскриптомики позволяет локализовать аберрации сетей и идентифицировать аномально регулируемые гены. Недавно высокопроизводительное scRNA-seq has было связано CRISPR/Cas9 мутагенезом, чтобы исследовать устойчивость сетей ( Jaitin et al., 2016; Dixi t et al., 2016; Adamson et al., 2016). Мы полагаем, что такая комбинация технологий предоставит важную информацию по регуляции дифференцировки клеток, клеточным коммуникациям, организации ткани и реакций на средовую изменчивость во время развития человека
. Conclusions Human organoids are manipula ble, genetically and otherwise, a feature once reserved for classical model systems such as yeast, worms, fli es, fish and mice. As a technology, however, in vitro organogenesis is still in its infancy, and in many cases it is unclear exactly what cell types are pres ent wi thin or ganoids and whet her each cell type can be created in a reproduc ible manner. scRNA-seq will help to address this uncertainty, providing a greater dep th of analysis of cell heterogene ity and reproducib ility. As protocols continue to evolve, human organoids are likely to come even closer to recapitulating bona fide human organogenesis in a predictable and reproducible way, making organoids a highly relevant system for understanding human development. We feel that quantitative single-cell transcriptomic approaches will provide impressive resolution of cell composition, lineage relationships, and gene network function within developing orga noids, and, together with other genomic approaches , will offer unprecedented insight into the mechanisms that underpin human organogenesis. Methods to analyze DNA, methylation, chromatin accessibility, nonmessenge rRNAs and proteins in single cells will further advance the field. The cost-per-cel l of many single-cell approaches is rapidly reducing and new methods are emerging that are relatively simple to implement in the lab. Hence, we believe that these technologies applied to human organoids represent a new direction in human developmental biology, and will help pave the way towards a better appreciation of what makes us uniquely human.
|