Посещений: 
с G БЕЛКОМ СЦЕПЛЕННЫЕ РЕЦЕПТОРЫ (GPCRs)
Механизмы передачи сигналов и избирательного агонизма
Mechanisms of signalling and biased agonism in G protein-coupled receptors Denise Wootten, Arthur Christopoulos, Maria Marti-Solano, M. Madan Babu & Patrick M. Sexton
 Nature Reviews Molecular Cell Biologyvolume 19, pages638–653 (2018)
| GPCRligGprot.htm
G protein-coupled receptors (GPCRs) are the largest group of cell surface receptors in humans that signal in response to diverse inputs and regulate a plethora of cellular processes. Hence, they constitute one of the primary drug target classes. Progress in our understanding of GPCR dynamics, activation and signalling has opened new possibilities for selective drug development. A key advancement has been provided by the concept of biased agonism, which describes the ability of ligands acting at the same GPCR to elicit distinct cellular signalling profiles by preferentially stabilizing different active conformational states of the receptor. Application of this concept raises the prospect of 'designer' biased agonists as optimized therapeutics with improved efficacy and/or reduced side-effect profiles. However, this application will require a detailed understanding of the spectrum of drug actions and a structural understanding of the drug-receptor interactions that drive distinct pharmacologies. The recent revolution in GPCR structural biology provides unprecedented insights into ligand binding, conformational dynamics and the control of signalling outcomes. These insights, together with new approaches to multi-dimensional analysis of drug action, are allowing refined classification of drugs according to their pharmacodynamic profiles, which can be linked to receptor structure and predictions of preclinical drug efficacy.

|
G protein-coupled receptors (GPCRs) являются крупнейшим семейством рецепторных белков клеточной поверхности у эукариот. У людей они кодируются свыше 800 индивидуальными генами и широко экспрессируются в разных тканях, где они контролируют широкий круг физиологических процессов. Всепроникающая роль GPCRs в физиологии человека возникает благодаря эволюционному разнообразию кодирующих последовательностей для 7 трансмембранных доменов (TMDs), которые формируют стержень рецептора, общий для всех GPCRs. На базе последовательностей и эволюционной консервации эти рецепторы подразделяются на подсемейства, которые включают подсемейства: класс A (rhodopsin-like), класс B1 (secretin receptor-like), класс B2 (адгезивные рецепторы), класс C (metabotropic glutamate receptor-like) и класс F (frizzled-like) , а также подсемейство taste 2 сенсорных рецепторов (GPCR Database). Разнообразие GPCR усиливается ещё больше за счет многочисленных механизмов, включая альтернативный сплайсинг, редактирование РНК, пост-трансляционные модификации и меж-белковые взаимодействия, которые изменяют как репертуар взаимодействующих лигандов, так и функциональные последствия активации рецептора 1. Такое разнообразие позволяет этим рецепторам распознавать и реагировать на огромное разнообразие лигандов, в пределах от фотонов, запахов, ионов, малых нейротрансмиттеров и малых нейромодулирующих пептидов до крупных гормональных пептидов и др. крупных белков с доменами, включая те, что участвуют в непосредственных межклеточных коммуникациях и в проникновении вирусов 2. Др. ключевой компонент многосторонности передачи сигналов GPCR - это широта внутриклеточных белков, которые они могут задействовать. Эти внутриклеточные партнеры включают многочисленные гетеротримерные G белки, которые служат в качестве канонических преобразующих (transducer) белков, а также в качестве регуляторных и каркасных белков, таких как arrestins, PDZ-домен содержащие каркасы и не-PDZ каркасы, такие как A kinase anchor proteins (AKAPs), которые инициируют или контролирую определенные паттерны передачи сигналов 3-7 (Fig. 1).
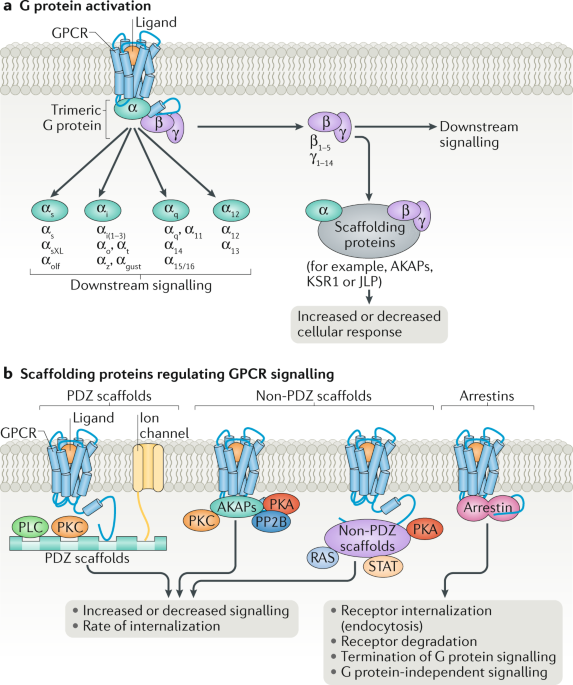 Fig. 1: Schematic illustration of GPCR signalling.
Fig. 1: Schematic illustration of GPCR signalling.
a | Canonical G protein-coupled receptor (GPCR) signalling occurs via coupling to heterotrimeric G proteins (Gα, Gβ and Gγ). Upon activation by a GPCR, the Gα and Gβγ subunits dissociate and can each activate downstream signalling. Gα proteins can be subdivided into four main families with different signalling properties. There are also multiple Gβ and Gγ subunits, which further diversifies signalling responses. Gα and Gβγ subunits can also associate with scaffolding proteins that regulate their signalling profiles. b| A schematic of GPCR scaffolding proteins that have key roles in the regulation of GPCR signalling and are also involved in forming higher order, tightly regulated signalling complexes, termed signalosomes. These scaffolds can be divided into three broad categories: PDZ scaffolds, which associate with the distal portions of GPCR carboxyl termini and can couple the GPCR to various signalling proteins such as kinases (for example, protein kinase C (PKC)), phospholipases (for example, phospholipase C (PLC)) and ion channels; non-PDZ scaffolds, such as A kinase anchor proteins (AKAPs), which bind to the cytoplasmic face of GPCRs and also associate with multiple signalling partners including kinases (for example, PKA and PKC), phosphatases (for example, serine/threonine-protein phosphatase 2B (PP2B)) and intracellularly localized receptors (such as inositol 1,4,5-triphosphate receptors (InsP3Rs) in the endoplasmic reticulum; not shown); and arrestins, which associate with many GPCRs, disrupting G protein-GPCR interactions and driving GPCR internalization via endocytosis, and act as scaffolds to facilitate multiple interactions between GPCRs and cytoplasmic signalling proteins in a G protein-independent manner. Of note, GPCRs themselves can serve as scaffolding proteins for other membrane proteins, including other GPCRs and receptor modifying proteins, as exemplified by receptor activity-modifying proteins (RAMPs) (not shown). JLP, JNK-associated leucine-zipper protein (also known as SPAG9); KSR1, kinase suppressor of RAS1; STAT, signal transducer and activator of transcription.
Чтобы осуществить передачу сигналов, GPCRs д. быть купированы с внутриклеточными преобразователями (transducers), такими как гетеротримерные G белки, которые формируются с помощью субъединиц Gα, Gβ и Gγ. У человека имеется 16 Gα, 5 Gβ и 13 Gγ субъединиц, которые могут быть скомбинированы, чтобы создать широкий круг гетеротримерных G белков. Каждая Gα субъединица может передавать сигналы независимо, тогда как Gβ субъединицы и Gγ субъединицы являются обязательными гетеродимерами, которые функционируют как одна единица(Gβγ?). 16 Gα субъединиц могут быть классифицированы в 4 основных Gα семейства (Gs, Gi/o, Gq/11 и G12/13), которые регулируют ключевые эффекторы (напр., adenylyl cyclase, phospholipase C, etc.) и генерацию вторичных мессенджеровs (напр., cAMP, Ca2+, inositol 1,4,5-triphosphate (Ins(1,4,5) P3), etc.), которые в свою очередь запускают определенные сигнальные каскады. Хорошо известно. что многочисленные определенные рецепторы могут быть купированы с одним и тем же Gα белком и что один и тот же рецептор может быть также купирован с более, чем одним Gα белком. Gβγ субъединицы обладают как регуляторной, так и сигнальной функцией, включая, напр., служение в качестве каркаса для рецепторных киназ и в качестве модуляторов для ионных каналов8. Совсем недавно наше понимание активации G белка продвинулось в направлении лиганд-зависимых эффектов на конформацию G белков, что связано с эффективностью передачи сигналов9,10. Успехи по клонированию и секвенированию и глобальные подходы по идентификации др. взаимодействий между GPCRи белками11,12 ещё больше расширили репертуар и/или сложность потенциальных последствий активации GPCR. Очень активно изучали партнеров по взаимодействию с GPCR, таких как arrestins, которые служат в качестве негативных регуляторных белков для передачи сигналов посредством G белков (путем блокирования активированных рецепторов посредством связывания гетеротримерных G белков (signalling desensitization) и посредством целенаправленного воздействия лигандом оккупированных GPCRs на эндоцитоз). Однако, они могут также функционировать как каркасы для инициации дополнительной передачи сигналов, преимущественно включая активацию разных MAPKs, таких как ERK13. Последняя часто наз. 'arrestin-зависимая, независимая от G белка' передача сигналов14, хотя степень, с которой такая передача сигналов может нуждаться в инициальном рекрутировании G белка, или может быть модулирована с помощью G белок-зависимой передачи сигналов, всё ещё исследуется15. Напр., посредством недавнего исследования потребности в G белке для arrestin-обеспечиваемой передачи сигналов было установлено, что передача сигналов (ERK фосфорилирование), обеспечиваемая с помощью arrestin, может нуждаться в G белке, но что arrestin-зависимая интернализация рецептора может быть достигнута в отсутствие функционального G белка15. В самом деле, использование arrestin GPCRs может происходить в отсутствие активации рецептора благодаря гетерологичному фосфорилированию с помощью вторичной мессенджер киназы16, и такое поведение д. быть понято, когда разрабатываются новые GPCR лиганды.
Принимая во внимание почти универсальную важность GPCRs в нормальном развитии и физиологии, неудивительно, что пертурбации с GPCRs и/или в их transducers может иметь большое значение в инициации прогрессировании болезни. Сегодня, ~30% разрешенных лекарств целенаправленно воздействуют на GPCRs, но эти лекарства действуют только на небольшой набор из репертуара GPCR 17,18, и имеется повышенный интерес к дальнейшей фармакологической эксплуатации этих белков 18. Тем не менее отсутствие ожидаемого клинического эффекта остается основной причиной недееспособности GPCR лекарств, это указывает на важные пробелы в нашем понимании передачи сигналов GPCR, в частности, их реакции на специфические лиганды. Препятствием к успешному фармакологическому целенаправленному воздействию на GPCRs является и болезнь-специфическая изменчивость в эффективности лекарств, которая возникает зависимым от пути передачи сигналов способом, от гетерогенности болезни, состояния прогрессирования болезни и изменчивости в поведении рецепторов, связанная с полиморфизмом рецепторных последовательностей внутри человеческой популяции 18,19. В последние годы наметился скачок в нашем понимании сложности механизмов и динамики функции GPCR, это обещает новые пути к идентификации и развитию новых GPCR лекарств и пониманию того, как они могут оптимально использоваться для терапевтических вмешательств. Среди наиболее продвинутых разработок является понимание, что GPCR лиганды могут обладать склонностью к агонизму, способности индивидуальных лигандов действовать на тот же самый рецепторов, чтобы инициировать разные клеточные исходы.
Complexity of GPCR signalling
Исторически GPCRs рассматривали как пассивные белки, необходимые для активации с помощью агониста, что позволяло им действовать, как избирательные каналы (conduits) между физиологическим (или фармакологическим ) лигандом и специфическим G белковым путем передачи. Соотв. подтипы GPCR всё ещё часто классифицируются в соответствии с их активирующим лигандом и их предпочтительным распознованием подсемейства G белков. Всё же большинство, если не все, GPCRs могут соединяться со многими преобразователями (transducer) и модуляторными белками, как следствие конформационной динамики, свойственной всем этим белкам 5,20,21 (Fig. 2).
 Fig. 2: Mechanisms of ligand-induced biased agonism.
Fig. 2: Mechanisms of ligand-induced biased agonism.
a | A schematic illustrating conformational dynamics occurring in G protein-coupled receptors (GPCRs). GPCRs can move between various inactive-like (R, R' and R'') and active-like (R* and R**) states. This can occur in the absence of ligand (Apo, black line); however, the energy barrier to achieving these states makes their occurrence a low probability. Addition of agonist or G protein (blue line) can decrease the energy required to reach active states, but full conformational change is favoured by the addition of both agonist and G protein (green line). b-d | Biased agonism can arise via multiple mechanisms. Distinct ligands induce different conformations within the receptor, resulting in different recruitment profiles for effector proteins such as G proteins and arrestins (part b). Ligand-induced receptor conformations can promote different conformational changes within scaffolding proteins such as arrestins, which in turn promotes activation of different downstream signalling pathways (for example, different MAPKs) (part c). Different ligands can induce distinct conformational rearrangements within G proteins that result in differences in the rate of GTP-GDP exchange. Ligands that induce a faster rate of GTP association (and hydrolysis) (top panel) allow quantitatively more G protein and downstream signalling events per unit of time than ligands that induce a slow rate of exchange (bottom panel) (part d). Pi, inorganic phosphate.
Conformational dynamics of GPCRs and diversity in GPCR signalling
GPCRs являются аллостерическими белками, которые делают возможными коммуникации между наружной средой и внутренностью клетки. Чтобы достичь этого они собирают множественные конформации, даже в отсутствие активирующих лигандов (apo состояние) (Fig. 2a). Эндогенные лиганды и лекарства изменяют конформационную динамику этих рецепторов, затрагивая временной и пространственный профили привлечения трансдукторов и регуляторных белков. Имеются доказательства, что такая конформационная пластичность может сильно влиять на реакции, которые вызываются данным лигандом4,5,22-24. Поведение лиганда может быть описано по двум ключевым параметрам: сродству (способности связываться) и эффективности (функциональные последствия связывания) (Box 1). Наше понимание сложности GPCR реакций переходит из упрощенной, линейной модели эффективности агониста - где все передачи сигналов пропорциональны - к модели, которая соответствует многомерности соединений рецептор-трансдуктор и активации трансдуктора, а также к концепции тенденциозного агонизма - способности индивидуальных взаимодействующих лигандов по-разному изменять паттерн нижестоящих клеточных реакций13,25 (Box 2).
Более того, для некоторых рецепторов, включая вирусные хемокиновые рецепторы гомологи GPCR US28 (ref.26), dopamine receptors27 и 5-hydroxytryptamine receptor 2C (5HT2C)28, лиганд-независимая передача сигналов (конституитивная активность), которая часто может быть демаскирована с помощью избыточной экспрессии или мутаций GPCR , также может обнаруживаться. Это указывает на то, что GPCRs выбираются, чтобы проявить широкий спектр внутренне присущих замалчиваний (quiescence) в противовес конституитивной активности в соответствии с функциональными нуждами. Биофизические и биохимические исследования подтвердили, что уровень конституитивной активности, обнаруживаемый индивидуальными рецепторами, связан с их конформационной динамикой и модулируется с помощью силы взаимодействий между аминокислотами внутри трансмембранного стержня рецептора, особенно между законсервированными полярными аминокислотами, которые формируют сети взаимодействий в основании рецептора5,21,29. Конституитивная активность GPCRs может быть модулирована как физиологически посредством изменений экспрессии рецептора или transducer, сплайсинга РНК и редактирования РНК (как отмечено для 5HT2C27), или посредством пост-трансляционных модификаций и при болезнях, когда альтерации этих механизмов, таких как с помощью мутаций active-conformation-stabilizing аминокислотных сетей или посредством альтераций клеточного окружения (изменений окружения мембран, pH, etc.), может происходить30,31. Активность GPCRs может также регулироваться прирожденно с примерами рецепторов, требующих индуцированной экспрессии (или шаперонов)32-34, чтобы достичь клеточной поверхности и рецепторов, которые подвергаются очень быстрому обороту, будучи динамически эндоцитозированными и преобразованными и отправлены обратно в мембрану35. Патологическая конституитивная активность вызывать болезни, как это наблюдается в случае конституитивно активного мутанта parathyroid hormone/parathyroid hormone-related peptide receptor (PTH1R) в случае Jansen's metaphyseal chondrodysplasia36 или при воздействии конституитивно активных мутантов внеклеточного Ca2+-sensing receptor CASR на потерю гомеостаза Ca2+37. Аберрантная конституитивная активность может также модифицировать болезнь , как в случае конституитивно активного metabotropic glutamate receptor 5 (mGluR5) при альтерациях головного мозга, наблюдаемых при нарушениях спектра аутизма38.
Очевидно, что GPCRs существуют во многих неактивных и активных конформациях, даже в apo состоянии (Fig. 2a). Исследования родопсина дали четкое определение множественных мета-стабильных конформационных состояний, варьирующих по продолжительности полу-жизни, которые изменяются во время управляемой протонами изомеризации ретинальной и рецепторной активации 39. Многие лиганд-независимые и лиганд-специфические состояния наблюдались также и для др. GPCRs 5,21.
Box 1 Understanding drug behaviour for clinical translation
To contextualize new advances in understanding structure-function relationships in G protein-coupled receptors (GPCRs), we must first consider how we describe and classify drug behaviour and define those components that are intrinsic to the bi-molecular interaction between ligand and receptor relative to those that may be cell-type-specific, organ-specific or even disease-specific (so-called system-dependent parameters).
Modelling signalling output
In its simplest form, the action of a drug can be separated into two key parameters: the ability of a drug to bind to the receptor (affinity) and its ability to trigger a cellular response upon binding (efficacy). A common pharmacological model for describing drug action at the receptor and cellular level is the operational model of agonism178, which ascribes a functional affinity and efficacy value to an observed response. This was originally applied to quantification of physiological measures, such as changes in whole organ responses, with the functional affinity value (KA) representing a macroscopic composite of all true microscopic affinities for each physical ligand-receptor-transducer complex making up the conformational ensemble. The efficacy parameter (?) in the operational model is a composite measure subsuming: the strength of interaction between each ligand-receptor complex that determines receptor coupling to a transducer protein to initiate a cellular signalling stimulus; the efficiency with which the stimulus is processed by cellular signalling pathways; and the total number of receptors mediating the observed response. By contrast, clinical efficacy is the observed, whole body outcome of integrated cellular responses across all target tissues upon administration of any pharmacological agent. At the most fundamental level, differences in drug behaviour are driven by the distinct chemical interactions between ligand and receptor, which determine the conformational sampling of the ligand-receptor complex to influence both transducer and regulatory protein interaction and transducer activation. This behaviour is independent of cellular context, and quantification of individual pathway efficacies in a chosen cell type can allow phenotypic clustering of drug chemotypes (see also below). Nonetheless, such surrogate measures of drug behaviour will be distinct from those displayed in native context, as each cell type will exhibit a unique level of expression, repertoire of receptors and composition of transducer, scaffolding and regulatory proteins that combine to determine cellular and tissue response. Moreover, this response can be further diversified by disease-specific changes to the proteins and the cellular environment. Successful drug discovery and development requires both an understanding of the full spectrum of drug behaviour and the ability to predict those properties in a manner that can bridge preclinical and clinical efficacy.
Overcoming translational barriers
Knowing that we have an incomplete understanding of the cellular consequence of GPCR drug action, how can we overcome the barrier of their pharmacological diversity? Observed bias can, and does, change with time, and it varies according to the breadth of end points used to analyse drug action. An increasingly popular approach is broad assaying of drug behaviour to cluster compounds into functional chemotypes66,151,179-182. The most advanced of these types of approach can interrogate over 30 end points of GPCR function, including kinetic measurement of effects on G protein recruitment and activation, signalling via second messengers and regulatory kinases and alterations to receptor trafficking. The functional chemotypes determined with these methods180,183 can now be linked to clinical drug behaviour (M. Bouvier, personal communication). Incorporating a minimum panel of diverse measures that can define a meaningful functional chemotype during compound validation, hit-to-lead and candidate nomination pathways that occur within a drug discovery pipeline may improve preclinical to clinical translation through greater pharmacological understanding of the drug leads entering into trials. Most importantly, though, insights from structural studies are now leading to the design of compounds with specific efficacies72, and solution of new structures of these designer ligands in complex with target receptors, combined with broad cellular assessment of receptor function, will provide increasing insight into the interactions that selectively alter receptor dynamics, leading to desired signalling profiles.
Box 2 Ligand classification, pluridimensionality of signalling and biased agonism
Ligands can be classified according to their binding site on the receptor, with the binding site of the canonical endogenous agonist termed the orthosteric site and binding sites that are topographically distinct from the orthosteric site termed allosteric sites. In some cases, orthosteric binding sites and allosteric binding sites can reside in close proximity, with ligands that can concomitantly bridge these two sites termed bitopic ligands184. Major outcomes from the increasing number of G protein-coupled receptor (GPCR) structures is recognition of the diversity of location of orthosteric and allosteric binding sites (with the location of these sites interchangeable across some receptor classes) and the observation that most, if not all, GPCRs possess at least one allosteric site185. Furthermore, allosteric ligands can be highly diverse, including Na+, which is a negative allosteric modulator of many class A GPCRs, classic small molecule compounds, antibodies, lipids and lipidated peptides (for example, pepducins)185,186. When used as drugs, allosteric ligands can possess the same spectrum of pharmacological activity as orthosteric ligands, but they establish distinct chemical interactions with the receptor. As allosteric ligands can bind simultaneously with orthosteric ligands, they can also alter the pharmacology of the latter, providing novel therapeutic opportunities (reviewed extensively elsewhere184-186). In effect, when bound to an allosteric ligand, the receptor-ligand complex can be viewed as a novel receptor with respect to how an orthosteric agonist propagates activation-associated conformational changes, and not surprisingly, alteration to the profile of transducer engagement is a common feature of many allosteric modulators.
An individual ligand can be typically classified as a full agonist, partial agonist, neutral antagonist or inverse (full or partial) agonist. The changes in receptor function arise from the specific chemical interactions that each ligand establishes with its target receptor, which impact the conformational ensemble of the GPCR, shifting the states sampled and the rate of interchange between conformational states. The efficiency with which the resulting conformational ensemble subsequently engages with or disengages from transducer-effector pathways thus leads to the aforementioned traditional phenotypic drug classifications. However, hitherto unappreciated differences in both the nature and the strength of chemical interaction between distinct ligands and an individual receptor can also drive changes in ligand residency and in transducer and/or regulator engagement that can vary for every downstream pathway. This is often referred to as 'pluridimensional signalling' and has led to the discovery of biased agonists as drugs that differentially promote this phenomenon. Although such differences in receptor-ligand complex conformations can be biophysically measured, these measurements are technically difficult and are often restricted to measurement of distance changes between a single pair of residues within the receptor.
In practice, the identification of biased agonism occurs via functional measures in cells but can be determined only when multiple (at least two) signalling end points are measured. Moreover, changes to signalling profiles can be subtle, requiring both depth of cellular interrogation and quantitative methods for measurement of relative agonist efficacy across all pathways. Because assessment of signalling bias is based on phenomenological readouts, the result can vary greatly according to cellular background and the nature of the response. Furthermore, biased agonism is always a relative term that is meaningful only when described relative to any differences from a reference agonist that has been assessed in parallel in the same assay and cell type. Terms such as unbiased ligand or balanced ligand, when used to describe a ligand with equivalent potency and/or efficacy in two different assays, can be problematic when system-dependent end points are measured (for example, cAMP accumulation versus arrestin recruitment), as alterations to the system background can change such relative potencies (for example, increased expression of G protein or altered expression of a GPCR kinase, which is linked to efficiency of arrestin recruitment). Thus, while biased agonism can be demonstrated from only two measures of cellular response, for example, G protein-mediated signalling versus arrestin-mediated signalling, this can provide only a very limited understanding of the potential pharmacologies of studied drugs. Consequently, if the two end points, even if different, are not the only (or worse, are not directly related to) therapeutically relevant signalling outputs, clinical translation will remain problematic. |
An evolving view of ligand pharmacology
Целью огромного большинства фармакоцевтических открытий и программ разработок является идентификация лекарств, которые или блокируют, или способствуют активации рецепторов. Для запуска активации индивидуальный лиганд обычно может быть классифицирован как полный агонист, частичный агонист, нейтральный антогонист или обратный (полный или частичный) агонист (Box 2). Различия как по силе, так и типу химических взаимодействий между разными лигандами и индивидуальным GPCR могут затрагивать время нахождения лиганда, управление изменениями в конформации рецептора и детерминации привлечения трансдуцера и/или регулятора, влияющих на тенденциозность агонизма.
Поскольку существовало общепринятое соглашение, что фундаментальной основой для тенденциозности агонизма являются лиганд-специфические изменения в конформации GPCR 40-42, вплоть до недавнего времени, классическая точка зрения заключалась в том, что дифференциальные передачи сигналов с помощью определенного лиганда (и отклонения в эффективности лекарств) возникают из-за различий в эффективности рекрутирования разных трансдуцеров на специфическую для лиганд-рецептор комплекса конформацию (Fig. 2). Новые исследования, использование bioluminescence resonance energy transfer (BRET)-based и fluorescence resonance energy transfer (FRET)-based биосенсоры конформации, внедренные внутрь субдоменов специфических G белков 9,10,43 или arrestins 44,45, показали, что конформационные отличия комплексов лиганд-рецептор распространяются также на трансдукторы (transducers). Для arrestins, такие конформационные изменения влияют на взаимодействие с потенциальными каркасными партнерами. Для G белков эти изменения влияют на величину связывания ГТФ, также как и на пребывание G белка в комплексах, связанных с GPCR, таким образом оборот G белка модулирует активацию своих нижестоящих сигнальных мишеней (Fig. 2). Более того, природа взаимодействия GPCR-arrestin, по крайней мере, частично зависит от фосфорилирования законсервированного мотива на рецепторе с помощью или GPCR kinases (GRKs) или вторичных мессенджер киназ 46. Накапливаются доказательства, что индивидуальные лиганды могут индуцировать специфические паттерны фосфорилирования рецепторов (обозначаются как штрих-коды фосфорилирования 47), сцепленных со специфическими поставками GRK 13,48 которые контролируют как силу взаимодействия аррестина с GPCR, так и последующее рекрутирование нижестоящих эффекторов 13,46-52.
Structural basis of GPCR signalling
Успехи в определении структуры GPCR и в биофизических измерениях динамики GPCR сегодня предоставляют ключевую информацию о том, как GPCRs активируются с помощью как соотв. лигандов, так и фармакологических агентов. Хотя детали варьируют между классами GPCR и даже между близко родственными рецепторами, но распространение конформационных изменений, возникающих от привлечения агонистов, сходятся на внутриклеточной стороне рецептора и приводят к законсервированному перемещению наружу трансмембранной спирали 6 (TM6) это делает возможным рекрутирование и активацию transducer. Тем не менее, несмотря на законсервированные механизмы привлечения трансдуцеров, нижестоящаяя передача сигналов от GPCRs может существенно варьировать в зависимости от контекста.
Class A GPCRs
Класс A GPCRs является наиболее многочисленным подсемейством GPCR и это отражается на количестве уникальных структур рецепторов, созданных для членов этого класса. Несмотря на огромное разнообразие их размеров и архитектуры 53, детальный анализ некоторых структур GPCR выявил общие, законсервированные, не-ковалентные контакты между эквивалентными рецепторными остатками в TMD, обозначаемыми молекулярными сигнатурами, которые сцеплены с молчащими рецепторами и переходом к активации (Fig. 3). Установлено существование лиганд-связывающей люльки 54. Хотя структуры многочисленных рецепторов известны, существует очень немного рецепторов, для которых структуры комплекса agonist-receptor-transducer (или миметик трансдуцера) и неактивного связанного с антагонистом или наоборот связанное с агонистом состояние известны. Рецепторы с известными этими двумя состояниями включают rhodopsin, β2-adrenergic receptor (β2-AR), muscarinic acetylcholine receptor M2 (mAChR), adenosine receptor A2A, µ-type opioid receptor (µ-OR) и κ-type opioid receptor (κ-OR) 5. Существуют общие свойства, которые могут определяться этими структурами. Напр., внеклеточная сторона рецептора подвергается контракции после связывания агониста и эта контракция аллостерически связана с открытием сайта, связывающего transducer. Более того, этот аллостерический механизм является реципрокным, т.е. работает и в противоположном направлении, после связывания внутриклеточного трансдуцера происходит закрытие лиганд-связывающего сайта 55.
 Fig. 3: Conserved residue contact networks between class A GPCRs and G proteins.
Fig. 3: Conserved residue contact networks between class A GPCRs and G proteins.
The G protein-coupled receptor (GPCR) and the G protein have been depicted in grey in cartoon and surface representations. Binding of ligands near the ligand-binding cradle in the receptor triggers distinct conformational changes in different receptor regions (schematically represented as black circles). These changes converge near the effector-binding region of the receptor through a conserved rewiring of non-covalent contacts among key receptor residues (represented as circles and labelled according to GPCR Database nomenclature. Engagement of these residues exposes receptor regions capable of recognizing a conserved G protein selectivity barcode (a pattern of amino acids specific to a given G protein; shown as a barcode logo). Binding to the receptor triggers a universal G protein allosteric mechanism that uncouples interactions between the α1 helix (H1), the α5 helix (H5) and the GDP-binding site of the Gα subunit and leads to GDP release. This allows GDP exchange for GTP and activation of the G protein. Connections between residues reflect inactivating (orange lines) and activating (green lines) contacts between them. Green dashed lines indicate that activation signals do not occur through direct contact. Анализ не-ковалентных контактов между эквивалентными остатками в доступных структурах в активной и неактивной конформации, показал, что несмотря на разнообразие конформационных изменений вблизи региона связывания лиганда между рецепторами, изменения сходятся вблизи региона связи с G белком (Fig. 3). Такая конвергенция обеспечивается с помощью сильно законсервированной структурной перестройки остатков контактов между TM3, TM6 и TM7, которые экспозируют остатки для контакта с G белком56. Это делает возможным взаимодействия с субдоменами Gα субъединицы, включая С-терминальную, α5 спираль, которая вставлена глубоко внутрь рецептора, как это видно на структурах в активном состоянии. Эти взаимодействия способствуют обмену нуклеотидами и позволяют активировать G белок. Такая конвергенция возможно может объяснить, как конформационные изменения, инициируемые лигандами, структурно очень разнообразными, позволяют GPCRs связывать широкий репертуар трансдукторов.
Ясно, что реорганизация рецепторов в полностью активное состояние не связана с присоединением только агониста, но и, скорее всего, отражает изменения динамики рецепторов, которая управляет как агонистом, так и трансдуктором5,21. Эта реорганизация иногда обозначается как 'loose allosteric coupling' и базируется на концепции, что рецептор может использовать множественные неактивные, промежуточные и похожие на активные состояния в отсутствие связи с лигандом (Fig. 2a). Такая потеря аллостерического соединения может быть видима при молекулярных динамических стимуляциях, при которых исследуется стабильность активного комплекса рецептор-агонист вследствие удаления G белка, как это иллюстрируется на структуре β2-AR-Gs. Эти симуляции показали, что рецептор может возвращаться к конформации, похожей на неактивную, даже в присутствии агониста57. Те не менее, множественные потенциальные мета-стабильные конформации были получены во время стимуляции, в частности, конформации, ассоциированные с изменениями в TM7, когда TM6 всё ещё оставался в похожей на активную конформации. Это согласуется с NMR исследованиями, выявившими сходные различия в позиционировании TM5 и TM7, но не обязательно в TM6, в состояниях β2-AR, предпочитаемых агонистами58,59 и с идеей, что взаимодействия с трансдуцерами необходимы для приобретения рецептором полностью активного состояния58,59. Недавние данные подтверждают, что G белки также могут формировать не-функциональные взаимодействия с рецепторами, которые могут быть преимущественно рецепторами для активации60,61. Такие данные также предоставляют доказательства, что помимо законсервированного глубокого связывания α5 спирали Gα субъединицы в стержне рецепт ора, др. важные взаимодействия между рецептором и G белком могут быть установлены. Тем не менее, это законсервированное, глубокое взаимодействие между GPCR и G белком, скорее всего, необходимо для активации G белка, т.к. оно вносит вклад в аллостерические конфорамационные изменения, необходимые для полного обмена нуклеотидами62,63.
Расширение доступности к известным структурам рецепторов и их комплексам с сигнальными трансдукторами позволит также провести более системный анализ купирования рецептора с трансдуктором и как нижестоящие G белки активируются. Анализ рецепторов, связанных и не связанных с G белками, вместе с детальным исследованием последствия консервации среди Gα субъединиц поможет выяснить универсальный аллостерический механизм активации Gα с помощью GPCRs62 (Fig. 3). Далее это было облегчено с помощью разработки системы < a href="https://www.mrc-lmb.cam.ac.uk/CGN/"> common G? subunit numbering (CGN) , которая позволила идентификацию эквивалентных остатков среди разных G белков62. Gα белки содержать домен RAS (назван из-за его гомологии малому G белку RAS). Короткие сегменты внутри этого домена подвергаются переходам от беспорядка к порядку как часть механизма активации белка G (первоначально описана для активации RAS), и эти изменения могут предоставить механизм, который совместим с путями консервативной аллостерической активации и избирательным связыванием G белка со специфическими типами рецепторов. Детальнее, после рекрутирования рецептора С-терминальная часть α5 спирали в Gα субъединице принимает более упорядоченную спиральную структуру, которая необходима для экспозиции остатков, ответственных за крепкое специфическое связывание разных Gα подтипов с GPCR. Молекулярная динамика симуляций показывает, что удаление ГДФ из Gα субъединиц достаточно, чтобы позволить это спиральное упорядочивание α5 спирали63 и подтверждает, что высвобождение нуклеотида предшествует полному задействованию G белка рецептором. В самом деле, в неактивном G белке ГДФ находится в непосредственном контакте с α5 спиралью, а также с α1 спиралью, являясь аллостерически связанным с высвобождением нуклеотида путем усиления гибкости, возникающего в результате перехода от беспорядка к порядку в α5 спирали, описанного выше. Т.о., рекрутирование G белка на GPCR может управлять высвобождением ГДФ, обусловленным с помощью аллостерических эффектов со стороны рецептора, и это, в свою очередь, может позволить последующее полное задействование рецептора и G белка путем перестройки α5 спирали. Это может потенциально объяснить, как система GPCR-Gα может быть столь разнообразной при механизме законсервированной распространенной аллостерической активации среди разных G белков62. Недавнее исследование также установило структурное объяснение, почему определенные рецепторы обладают способностью соединяться с одними и теми же самыми типами G белков, но не с др. Детализация последовательностей и структурный анализ человеческих GPCRs и G белков выявил существование паттернов аминокислот, наз. G белковыми избирательными штрих-кодами, на каждом из 16 человеческих Gα белков, которые могут быть распознаны за счет определенных регионов среди приблизительно 800 человеческих рецепторов. Важно, что некоторые позиции в штрих-коде сильно законсервированы, тогда как др. являются уникальными для индивидуальных G белков. В то время как сильно законсервированные позиции в избирательности штрих-кода позволяют рецепторам соединяться с и активировать G белки сходным способом, уникальные позиции распознаются с помощью специфических рецепторов благодаря определенным остаткам и только некоторые штрих-коды могут быть распознаны любым данным рецептором. Эту ситуацию можно сравнить со сценарием, имеющим множественные ключи (рецепторы), открывающими один и тот же замок (G белок), используя не-идентичные паттерны. Более того, было показано, что изучение эволюционной истории GPCRs позволяет идентифицировать эти избирательно детерминирующие остатки на рецепторе20.
Помимо привлечения G белка, по крайней мере, для класса A GPCRs, накапливаются доказательства, что конформация TM7 и особенно того консервативного мотива NPXXY (in single letter amino acid code, where X is any amino acid) может вносить вклад в эффективность связывания и активации arrestin и, следовательно, в наблюдаемую передачу сигналов между агонистами21,64-66. Это подтверждается спектроскопическим исследованием β2-AR в комплексе с агонистом с дивергентной фармакологией, особенно тех, то активируют как G белок, так и передачу сигналов arrestin и тех, обнаруживающих склонность к рекрутированию arrestin67. В этом исследовании в отсутствии трансдуцера, G protein-компетентного и arrestin-компетентного лигандов изменяется конформация как TM6, так и TM7, в то время как те, что обнаруживают ограниченное использование G белка и предпочитают arrestin-обеспечиваемую передачу сигналов, преимущественно изменяют только конформацию TM7. Сходным образом, дифференциальное рекрутирование arrestin на κ-OR в противовес µ-OR с помощью лиганда 3-iodobenzoyl naltrexamine (IBNtxA) может быть изменено мутациями ключевого остатка в TM7 (Y/W7.35; single amino acid code numbered according to the Ballesteros and Weinstein class A numbering scheme68). Эта мутация индуцирует незначительные изменения в ориентации лиганда в отношении двух рецепторов, подтверждая, что это различие в позиции, связывающей лиганд, и в силе химических связей между лигандом и рецептором, достаточны для изменения ключевых компонентов внутримолекулярных аллостерических сетей внутри рецептора, которые управляют избирательной передачей сигналов69. Роль конформационных изменений TM7 была также предположена для дифференциального привлечения трансдуцеров с помощью 5HT1B и 5HT2B, стимулируемого с помощью разнородного агониста ergotamine. Этот лиганд активирует пути как G белка, так и arrestin ниже 5HT1B и прежде всего активирует arrestin-обеспечиваемую передачу сигналов ниже 5HT2B. В этом случае ключевым различием оказывалось конформационное изменение TM7 (ref.65). Дополнительная информация о механизмах управления рекрутирования arrestin получена при исследовании кристаллической структуры 5HT2B, связанного с lysergic acid diethylamide (LSD), при этом единичная точечная мутация во внеклеточной петле 2 (ECL2; срелдиняющей TM4 и TM5), которая снижала взаимодействие с LSD и увеличивала норму (off-rate) лиганда, заметно ослабляла рекрутирование arrestin 70,71. Интересно, что комбинация этих знаний с предыдущей информацией о склонности агонизма dopaminergic рецепторов, позволила идентифицировать ряд фармакологических соединений, обнаруживающих склонность к β-arrestin72.
Известно, что дифференциально фосфорилированные С-терминальные пептиды, которые подражают фосфорилированным С-терминальным хвостам рецептора, м. индуцировать определенные конформации arrestin в регионах, ответственных за образование белкового каркаса73,74. Т. о., дополнительный механизм предпочтительности может быть связан со специфичными для лиганда различиями в соединении комплекса рецептор-лиганд с разными киназами, это, в свою очередь, меняет паттерны фосфорилирования С-конца рецептора (штрих-код фосфорилирования). Недавняя работа пролила дополнительный свет на сложные взаимоотношения между фосфорилированием С-терминальных хвостов рецептора и рекрутированием arrestin, это доказывает аллостерическую модуляцию стержня рецептора с помощью phospho-carboxy-терминального хвоста, который регулирует связывание arrestin со стержнем β2-AR52. Многие GRKs нуждаются во взаимодействии с βγ субъединицей G белка, это позволяет объяснить, почему передача сигналов arrestin требует соединения G белка с рецептором. Очевидно, что связывание того же самого фосфопептида с др. классом A GPCRs, как было установлено, выявляет разные эффекты на использование arrestin с рецептором75. Эти различия в привлечении arrestin с помощью фосфорилированных рецепторов может быть объяснено тем фактом, что существует, по крайней мере, две основные конформации при взаимодействии arrestin-GPCR : конформация хвоста и конформация сердцевины, которые отличаются для разных пар arrestin-GPCR. Конформация хвоста возникает в результате взаимодействия arrestin с фосфорилированным С-терминальным хвостом GPCR76 и это совместимо с сопутствующим взаимодействием GPCR-G белок77. При конформации стержня помимо взаимодействия с хвостом, arrestin осуществляет взаимодействия со стержнем рецептора посредством finger-loop домена, как это показано для кристаллической структуры rhodopsin-arrestin76. Эти две конформации были связаны со специфическими функциями arrestins (arrestin при конформации хвоста остается функциональным в интернализации GPCR и в некоторых формах передачи сигналов, тогда как desensitization рецептора объясняют конформацией стержневой части рецептора)49. Хорошо бы понять степень, с которой С-концевой хвост рецептора (или внутриклеточные петли) подвергаются структурной реорганизации после фосфорилирования, чтаобы повлиять на конформации рецептора и трансдуцера. В недавней работе с минимально модифицированным классом B GPCRs43,78,79 было показано, что С-терминальный хвост рецептора конформационно динамичен, по крайней мере, в состоянии связи с G белком.
Большинство исследований связи структуры белка со тенденциозным агонизмом было ограничено преимущественно G белковым трансдуцером как мишенью для рецептора и рекрутированием arrestin и это ограничивает на it понимание спектра лигандами вызываемых переключений конформации, которые вносят вклад в тенденциозную передачу сигналов. Это привлекло существенное внимание к разработке multiplexed подходов по исследованию изменений функции GPCR (Box 1). В недавнем исследовании, комбинирующем анализ эволюционного отслеживания для предсказания и в дальнейшем для мутирования 28 аминокислот β2-AR, который может участвовать в определенных конформационных сетях, управляющих тенденциозной передачей сигналов 66. Путем оценки конституитивного и агонистом вызываемого рекрутирования G белка, активации G белка, рекрутирования arrestin и эндоцитоза рецептора, эта работа предоставила дополнительные доказательства исключительной важности консервативных мотивов в TM7 для дифференциальной способности β2-AR привлекать разные трансдуцеры
Class B GPCRs
Класс A GPCRs обнаруживает значительные различия в размере и архитектуре своих N-терминальных внеклеточных доменов (ECDs). Так, класса B1 пептидных гормональных GPCRs обладает ECDs, которые приблизительно длиной в 100-150 аминокислот и образуют законсервированную 3D укладку, которая связывает С-терминальный сегмент активирующего пептидного лиганда 80.недавние структурные исследования расширили наше понимание активации класса B1 GPCRs и предоставило первую информацию о механизмах, которые вносят вклад в тенденциозный агонизм. Эти рецепторы поддерживаются в неактивном состоянии серией законсервированных, полярных, связанных водородными мостиками сетями аминокислот в основании рецептора, которые наблюдаются в структурах с неактивным состоянием изолированных TMD 81-84 (Fig. 4a). Кроме того, имеются доказательства, что взаимодействия между доменами N--терминального ECD и трансмембранного рецепторного стержня могут удерживать рецептор в молчании 85,86.
 Fig. 4: Conformational changes in class B and class C GPCRs required for G protein coupling.
Fig. 4: Conformational changes in class B and class C GPCRs required for G protein coupling.
a | The inactive homology model of glucagon-like peptide 1 receptor (GLP-1R), based on the inactive structure of the related glucagon receptor (left) and active, exendin-P5-bound GLP-1R structure40 (right), highlighting key amino acid side chains that undergo reorganization during activation transition. These are grouped according to their function and displayed in distinct colours. Prominently, polar, hydrogen-bonded interactions at the base of the receptor are broken or reorganized in the active state. Clear differences in the organization of the hydrophobic network can also be observed between the inactive and active receptor structure. Class B G protein-coupled receptor (GPCR) activation is also associated with conserved changes in the orientation of transmembrane (TM) helices, an outward movement of the extracellular ends of TM6 and TM7 and an inward movement of the extracellular end of TM1. These changes culminate in an outward movement of TM6 at the cytosolic side, which supports accommodation of the transducer. b | A schematic of class C GPCR activation. These receptors are functionally dimeric (homodimer form shown) and bind ligand (green circle) via the large extracellular 'venus fly trap' (VFT) domain. Notably, the VFT domain spontaneously oscillates between open and closed conformations. Agonist binding leads to conformational changes in the VFT domain that support the closed conformation of the VFT, and these alterations are linked to repositioning of the TM domain bundles from the two receptor subunits towards each other. As a result, these transitions induce an asymmetric conformational change in one protomer that enables G protein binding. The structural details behind these transitions are not known. The purple circles indicate cysteines in the TM bundle or cysteine-rich linker domain. H8, helix 8.
В последние годы выявлены множественные структуры, содержащие как ECD, так и трансмембранную стержневую часть, включая рецепторные комплексы с ингибирующими антителами87 и частичными агонистами88,89, а также три структуры рецепторов полной длины с комплексе как с пептидным агонистом, так и гетертримерными Gs белками43,78,79, делающими этот самый богатый класс рецепторов полностью активными, связанными с G белком структурами. Сравнение TMDs структур в неактивном состоянии со структурами agonist-receptor-G protein у calcitonin receptor (CTR)78 и glucagon-like peptide 1 (GLP-1) рецептора (GLP-1R)43,79 выявило общие крупно-масштабные изменения в архитектуре трансмембранного стержня после связи с пептидным агонистом. Они включали заметное изгибание и перемещение кнаружи внеклеточных концов TM6 и TM7 и перемещение внутрь внеклеточной вершины TM1. Эти изменения уникальны для класса B GPCRs, но как наблюдалось и для класса A GPCRs, они преобразуются в перемещение кнаружи TM6 в основании рецептора. Это событие законсервировано среди всех классов GPCR и делает возможной accommodation α5 спирали Gα. Такое крупномасштабное изменение происходит вокруг консервативных мотивов класса B1, включая Pro6.47b-X-X-Gly6.50b (superscript numbers refer to the class B GPCR transmembrane core numbering system90) в TM6, Gly7.50b в TM7 и Gly1.46b в TM1.
Сравнение полностью активного с G белком связанного GLP-1R с недавней структурой родственной glucagon receptor (GCGR), связанному с частичным агонистом, в отсутствие G белка88, может предоставить информацию о последовательности конформационных изменений, которые реобходимы для привлечения G белка для класса B GPCRs. В структуре partial agonist-GCGR рецептор обладает только субнабором изменений трансмембранного стержня, наблюдаемых в структуре связанной с G белком. В частности, в то время как происходит реорганизация ECL1 и ECL2 и перемещение кнаружи TM6, чтобы приспособиться к связыванию пептида, TM1 и TM7 не искривлены, а основание рецептора перекрывается со структурой, характерной для неактивных рецепторов. Поскольку мы пока не понимает последовательности событий, управляющих полной активацией рецептора, то структура GLP-1R, связанная с 11-mer пептидным агонистом89 и аллостерическим ингибитором84 предоставляет дополнительную информацию, как минимальные конформационные изменения, делают возможным приспособление к полностью активному состоянию. Каждая из этих последних структур обладает ключевыми свойствами полностью активного состояния в организации внеклеточного сегмента стержня рецептора, включая реорганизацию ECL2, перемещение кнаружи TM6 и TM7 и перемещение внутрь TM1. Обе структуры были модифицированы в ключевых трансмембранных сегментах, чтобы сделать возможной стабилизацию и кристаллизацию рецептора. В структуре GLP-1 R, связанной с ингибитором, дисульфидная связь была внесена между TM6 и TM5, это привело к разрыву взаимодействий, которые стабилизируют негативное состояние, указывая тем самым, что такое нарушение может быть существенным, чтобы управлять структурными изменениями в отсутствие др. ограничений. В 11-mer-агонистом-связанной структуре мотив Pro-X-X-Gly в TM6 был мутирован и это также может вносить вклад в имитирование полностью активного состояния верхней половины стержня рецептора. Вполне возможно, что существует скоординированная серия взаимодействий между пептидом и рецептором и между рецептором и G белком (или др. трансдуцером), которые являются аллостерически передаваемыми между внутриклеточными и внеклеточными доменами и делают возможной адаптацию к полностью активному состоянию. Эти переходы согласуются с рыхлым аллостерическим купированием, описанным выше для класса A GPCRs и прекрасно соответствует биофизическим наблюдениям конформационных изменений, ассоциированных с комплексами agonist-receptor, receptor-G protein и с полными комплексами agonist-receptor-G protein в классе A GPCRs5,21.
Современная общая модель для активации класса B GPCR может быть суммирована следующим образом: после инициального связывания пептидного агониста, происходит реорганизация рецепторных ECLs, которые приспосабливают связывание пептида к стержню рецептора и вносят вклад в распространение конформационных изменений, связанных как с активацией рецептора, так и тенденциозностью агонизма43,91,92. Внутри стержневой части рецептора происходит реорганизация законсервированной центральной сети полярных аминокислот, а мутационные данные подтверждают незначительную роль этой сети в склонности к пептид-специфической передаче сигналов93. Ниже этой сети в направлении внутриклеточной стороны рецептора имеются гидрофобные остатки, которые стабилизируют как неактивное, так и активное (скомплексованное с G белком) состояние, хотя эти процессы происходят за счет разных взаимодействий. Активация рецептора ассоциирует с разрывом ключевой полярной сети в основании рецептора; это необходимо для перемещения TM6 и взаимодействия с G белком43,78,79,94,95(Fig. 4a). Динамика, лежащая в основе этих конформационных изменений пока непонятна. Однако, мутационные данные подтверждают роль перераспределения консервативных, полярных или плотно упакованных малых аминокислот в этом переходе90. Недавняя структура GLP-1R, связанного с его главным физиологическим лигандом, GLP-1 (ref.79), или с тенденциозным агонистом exendin-P5 (ref.43) также предоставили новую информацию о структурной основе эффективности и тенденциозности передачи сигналов этим рецептором, показав ключевые отличия в конформации ECL3 и верхушки TM1 рецептора, когда он соединяется с разными лигандами.
Интересно, что все известные с G белком скомплексованные структуры существуют в мономерных формах. Однако, имеются серьёзные доказательства, что класс B1 GPCRs подвергается как гомодимеризации, так и гетеродимеризации и что это функционально важно, т.к. нарушение димеризации ассоциирует со снижением потенциала агониста для канонических Gs, купированных с рецептором 80. Димеризация также может вносить вклад в тенденциозные профили аногистов 96 и в изменения клеточных реакций на агонисты в отобранных гетеродимерных рецепторах класса B 97,98, хотя это не было ещё изучено тщательно. Ограниченные данные подтверждают, что рецепторы являются димерными после инициального взаимодействия с лигандом, это вносит вклад в связывание с высоким сродством, но эти димеры дестабилизируются после активации и взаимодействия с G белком, это может способствовать предпочтительному выделению комплексов мономерных рецепторов с G белком в структурных исследованиях 9,80,99.
Class C GPCRs
В отличие от многих др. семейств GPCR класс C GPCRs являются обязательными димерами и это критически связано с их активацией 100-102.Напр., GABA типа B рецептор является облигатным гетеродимером, при этом одна субъединица предоставляет место для связывания агониста, а др. предоставляет домен для связывания G белка 103. Класс C рецепторов содержит очень крупные внеклеточные домены, которые представлены структурно законсервированным модулем 'venus fly trap' (VFT), который действует как эндогенный лиганд-связывающий сайт и соединительный домен, который продолжается в TMD. VFT модули обладают изменчивой сложностью в зависимости от подтипа рецептора. Orthosteric связывание агониста с доменом VFT порождает конформационный сдвиг модуля VFT из открытого в закрытое состояние, это, в свою очередь, вызывает заметные изменения в позиции и ориентации TMDs внутри димера рецептора, что запускает связывание трансдуцера 104 (Fig. 4b). FRET исследования конформационной динамики в mGluRs показали, что VFT домены могут спонтанно осциллировать между покоящейся (открытой) и активной (закрытой) ориентацией и что агонисты разной эффективности по-разному изменяют это равновесие 104, это выступает в качестве механизма управления активацией рецептора. Кстати, пока недоступна структура полной длины рецептора и природа взаимодействий между TMDs димера, которые участвуют в активации рецептора, неясна. Однако, для mGluR2, образование цистеиновых дисульфидных мостиков между TM4-TM5 субъединицами димера предупреждает обусловленную агонистом активацию, тогда как поперечное связывание TM6-TM6 делает рецептор постоянно активным 101, это согласуется с реорганизацией димерного интерфейса как части механизма активации. Конечно, как позитивные, так и негативные модуляторы, соединяющиеся с TMD, были идентифицированы для большинства рецепторов класса C 105. В рецепторе полной длины большинство позитивных регуляторов лишено прирожденной действенности, хотя они могут активировать N-терминально укороченные рецепторы, лишенные ECD, указывая на то, что ECD вносит вклад в поддержание неактивного состояния. Тем не менее, такие модуляторы могут модифицировать профиль передачи сигналов orthosteric агониста, как это наблюдается в случае CASR 106. Это наблюдение показывает, что в классе C рецепторов, как и в др. классах GPCR, аллостерические модуляторы изменяют конформационные выборки рецепторов во время перехода к активации 80,107.
Local control of GPCR signalling
Наше понимание передачи сигналов GPCR также продвинулось вперед в отношении оценки важности локализации рецептора внутри клетки и роли клеточного окружения в каждой локации (Fig. 5). Этот локальный контроль может включать 'латеральную' аллостерию посредством взаимодействия с мембранными липидами и изменения в композиции липидов 108-113 и привлечение рецепторов в белковые комплексы сигналосом, которые ограничивают или изменяют взаимодействия эффекторов и регуляторов, воздействуя тем самым на рецепторы и передачу сигналов 4,114. Более того, локализация рецептора может также влиять на функциональную избирательность агонистов, отличающихся физико-химическими свойствами, посредством чего гидрофильные соединения нуждаются в экспрессии на клеточной поверхности рецепторов, чтобы действовать, но липофильные соединения могут достигать пространственно ограниченных рецепторов, расположенных внутри клетки, как это было продемонстрировано для β1-AR23.
 Fig. 5: Compartmentalization of signalling by GPCRs.
Fig. 5: Compartmentalization of signalling by GPCRs.
Signalling by G protein-coupled receptors (GPCRs) can be spatially (and temporally) compartmentalized to encode unique responses at the cellular level. This is facilitated by the formation of higher order protein complexes (signalosomes) around GPCRs and through the signalling of GPCRs at multiple locations within the cell - apart from the canonical signalling initiated at the plasma membrane, GPCRs can signal from endosomes after receptor internalization. They can also be activated by membrane-permeable ligands from intracellular organelles (including endosomes and the Golgi apparatus). The composition of the lipid bilayer can also influence GPCR signalling. For example, localization of receptors and their signalling partners into microdomains (such as caveolae or lipid rafts) can physically influence their conformation and signalling output through interactions with lipids and membrane proteins that are enriched in these domains (lateral allostery). Spatial and temporal dynamics can also be regulated by physical barriers, such as the cytoskeleton, and by biochemical buffering. An example of this latter process is the buffering of GPCR-dependent Gs-mediated cAMP production, whereby discretely positioned phosphodiesterases degrade cAMP, acting as local cAMP sinks. This buffering can occur at various sites within the cell, including at the plasma membrane and the endosomes, to control cAMP diffusion and to create discrete cAMP gradients within a cell.
Membrane composition
В то время как мы обычно обозначаем окружение рецепторов ка липидный бислой и полагаем. что плазматическая мембрана является ключевым сигнальным доменом, физиологические мембраны могут быть очень сложными, содержать до 1000 разного типа липидов 115,116, которые могут взаимодействовать непосредственно с рецепторами и могут также собираться в микродомены благодаря взаимодействиям с мембранными белками (наиболее распространены caveolins для формирования кавеол). Такие микродомены могут быть обогащены специфическими трансдуцерами, включая подтипы G белков 117. Различия в липидном составе также влияют на изогнутость мембраны, что, в свою очередь, может влиять на конформационную динамику GPCR 118. Очевидно, GPCRs могут быть динамически распределены между такими микродоменами, приводя к альтерациям в передаче сигналов и это может вносить вклад в наблюдаемые различия в поведении лекарств. Важным примером является µ-OR, т.к. компартментализация этого рецептора внутри определенных субдоменах плазматической мембраны, играет ключевую роль в отличающихся фармакологических реакциях на лекарства 24.
Receptor trafficking
GPCRs подвергаются циклам динамического переноса через разные клеточные компартменты. Этот аспект также регулируется с помощью зависимых от лиганда взаимодействий, как части десенсибилизации (desensitization) специфических сигнальных трансдуцеров и использования альтернативной передачи сигналов, а также подавления реакции рецепторов, что сопровождается целенаправленной деградацией рецепторов. Величина спонтанного и лиганд-зависимого трафика рецепторов между разными компартментами мембраны скоординирована с селективных взаимодействием рецепторов с репертуаром аппаратов сортировки и транспорта. Очевидно, что скорость доставки рецепторов обнаруживает значительную изменчивость среди GPCRs и может по-разному изменяться в ответ на разные лиганды 119-123. Эти события доставки контролируются с помощью конформационных выборок состояний рецепторов (и последующих событий, включая пост-трансляционные модификации, такие как фосфорилирование, palmitoylation и убиквитинирование) и, как было установлено, специфические концентрации лигандов, химиотип и время пребывания 119-123. Неудивительно, что даже в отсутствии внутриклеточной передачи сигналов, агонист-избирательные отличия в скорости и степени поставки рецепторов могут влиять на паттерны клеточной реакции и составлять компонент фармакологического профиля лекарства, который вносит вклад в наблюдаемую действенность клеток .
Assembly of signalosomes
Хотя мы знаем уже многие годы, что GPCRs и др. белки могут собираться в мульти-белковые комплексы и что это может влиять на наблюдаемую функцию рецепторов и клеточные реакции (изучено исключительно на передаче сигналов в нейрональных синапсах), наша оценка важности этих событий для общего представления, такого как тенденциозность агонизма, лишь недавно стала реальной4. Простейшая сборка единиц, представляющая рецепторные димеры или олигомеры, активно обсуждаются124,125. Стоит отметить, что гетеромерные ансамбли рецепторов может изменять привлечение трансдуцеров или поставку рецепторов в ответ на физиологические лиганды125,126 и что динамические изменения одного из партнеров рецептора могут вносить вклад или в болезнь или в лечение болезни. GPCRs могут также взаимодействовать с не-GPCR партнерами, чтобы изменить распознавание лиганда, привлечение и активацию трансдуцера или поставку рецептора. Сюда входит и взаимодействие GPCRs с каркасными белками, такими AKAPs и др. белками3-5,7,103, которые считаются каркасами для мульти-белковых ансамблей, которые осуществляют тонкую настройку передачи сигналов (see below). GPCRs могут также взаимодействовать с мембранными белками, такими как receptor activity-modifying proteins (RAMPs)33, melanocortin receptor-accessory proteins (MRAPs)34 и low-density lipoprotein (LDL) receptor-related proteins (LRP)127. RAMPs, это семейство из белков с тремя трансмембранными участками, вызывающих спектр эффектов, которые могут вызываться с помощью таких взаимодействующих белков. RAMPs первоначально были распознаны по их необходимой роли в доставке функциональных calcitonin receptor-like receptor (CALCRL)-RAMP1 гееродимеров (CGRP) или adrenomedullin receptors (CALCRL-RAMP2 или CALCRL-RAMP3 гетеродимеров) на клеточную поверхность, внося вклад как в извлечение CALCRL из аппарата Гольджи так и/или в эндоплазматический ретикулум и в формирование связывающего лиганд кармана этих рецепторов128. RAMPs теперь считаются широко распространенными партнерами GPCRs, включая и те из всех основных подсемейств32,33. Хотя они и не сильно необходимы для экспрессии на клеточной поверхности GPCRs, RAMPs могут менять регуляцию рецептора, специфичность лигандов и паттерны привлечение трансдуцеров33. Поскольку RAMP и др. взаимодействующие белки, динамически регулируются в нормальной физиологическом состоянии и при болезнях, то влияние этих партнеров GPCR на функцию рецепторов д. учитываться при поиске и разработке лекарств.
В целом сборка GPCRs в сигналосомы может предоставлять клеткам очень тонкий контроль над передачей сигналов и коррелирует с пространственно ограниченной передачей сигналов 114,129-132, с очень высокой чувствительностью реакции 133 и контролем специфичности присоединения трансдуцеров 103,134,135(Fig. 5). Это последнее свойство в частности необходимо учитывать при трансляции профилей тенденциозных агонистов, измеряемых в рецепторах, экспрессирующихся посредством рекомбинантной системы экспрессии белка в противовес той, что существует в клетках мишенях всего органа (здорового или больного). Более того, сборка GPCRs в комплексы сигналосом, скорее всего, изменяет их конформационные выборки, при этом обнаруживается потенциал таких взаимодействий, чтобы аллостерически регулировать связывание лиганда, нижестоящую передачу сигналов и др. регуляторные механизмы, как это наблюдается при взаимодействии RAMP-GPCR 33.
Physiological consequences of compartmentalized signalling
Потенциал GPCRs передачи сигналов из внутриклеточных компартментов традиционно связывают с arrestin-зависимой интернализацией рецепторов и рекрутированием независимых от G белка трансдуцеров на эндоцитозированные рецепторы. Однако, феномен, такой как сохранение активации G белок-зависимых путей, указывает на роль передачи сигналов G белками с мест вне плазматической мембраны136. Успехи в инструментарии по локализации компонентов сигнальных комплексов привели к детекции активации трансдуцеров в разных клеточных субдоменах и это указывает на передачу сигналов не только от эндосом после эндоцитоза, а также с мест, включая секреторные компартменты (эндоплазматический ретикулум и аппарат Гольджи), ядро и митохондрии137-142. Более того, эксперименты с избирательным вмешательством в передачу сигналов из внутриклеточных компартментов продемонстрировали. что эти передачи сигналов являются физиологически важными и могут в принципе стать терапевтическими мишенями, как, напр., в случае β2-AR141, NK-1 receptor (NK-1R; известен также как TACR1)143, CGRP144и PTH1R136,142. Интересно, что общение между рецепторами может управлять изменениями в компартментализации передачи сигналов, примером может служить временная активаци β2-AR, посредством Gβγ субъединиц, модулируя продолжительность передачи сигналов эндосомного PTH1R145.
Альтерации физиологически важных компартментализаций передач сигналов, скорее всего, будет вызывать болезни или модифицировать болезни, как это продемонстрировано для β-ARs при сердечной недостаточности 129,130,146 и для melanocortin receptor 4 (MC4-R) при некоторых генетических формах ожирения 134. Напр., пространственное ограничение β2-AR (а, следовательно, и передачи сигналов цАМФ) глубокими поперечными трубочками здоровых кардиомиоцитов нарушается у крыс, моделирующих хроническую сердечную недостаточность transverse, приводит к диффузной передаче патологических сигналов цАМФ 13. Более того, избирательная способность индивидуальных лигандов рецепторов, чтобы регулировать переходы рецепторов между компартментами (напр., между разными клеточными компартментами или микродоменами плазматической мембраны) могут вносить вклад в наблюдаемую тенденциозность профилей агонистов 24,147.
Translational considerations
Итак, упомянутые выше успехи структурной биологии GPCR внесли вклад в более детализированную картину влияния лиганда, рецептора и структурных детерминант сигнальных трансдуцеров на передачу сигналов и действенность лигандов. Важно, что эта структурная информация подчеркивает новые пути модулирования GPCRs более изящным способом, тем самым прокладывается путь для разработки новых химических биологических инструментов для определения функции рецепторов и, что более важно, для разработки нового поколения лекарственных средств.
Understanding the impact of ligand-receptor kinetics on receptor signalling
Большинство взаимодействий рецептор-трансдуцер и признаков активации трансдуцеров, описанных выше, имеет следствием кинетику взаимодействия лиганд-рецептор, расположение лиганда и последующую вероятность привлечения трансдуцера или регуляторного белка. Это является компонентом избирательного, би-молекулярного взаимодействия между индивидуальными лигандами и целями рецепторами и становится привлекательным фокусом для некоторых терапевтических программ разработки лекарств. Не удивительно, классификация наблюдаемых тенденциозных агонизмов может меняться в зависимости от временного разрешения функционального считывания передач сигналов и это также предоставляет потенциал рационализации для расхождений в классификации лигандов в разных лаб. 22. Расположение лигандов, в частности, становится областью внимания для понимания фармакологической дифференцировки соединений и кинетики лиганд-рецептор взаимодействий, связанных с действенностью передачи сигналов, плейотропией (включая рекрутирование arrestin) и клинической эффективностью соединений 22,71,148-150.
Identification and progression of biased agonists in drug pipelines
Ключевым уроком из всё увеличивающейся детально молекулярной информации по поведению GPCR и последствиям их взаимодействия с лекарствами, является то, что передача сигналов GPCR является сложной и что обнаружение и разработка лекарств для этих рецепторов затруднительна. Тенденциозный агонизм может быть выявлен с помощью минимум двух измерений клеточных функций (т.е. путем мониторинга активности двух независимых исходов от передачи сигналов), но понимание природы тенденциозности передачи сигналов лекарств и ей вклад в модулирование болезни требует понимания путей, связанных как с желательной терапевтической эффективностью, так и потенциальными побочными эффектами 151. На сегодня мы имеем лишь частичное понимание передачи терапевтических сигналов. Имеются чрезвычайно сложные исследования с использованием мышей с химиогенетическими модификациями рецепторов или со светом модулированных рецепторов (оптогенетика), чтобы специфически изменять передачу сигналов рецепторов и выявить ключевые физиологические и патофизиологические реакции на такие измененные передачи сигналов 142-155. Такого типа исследования могут определить ключевые компоненты передачи сигналов, которые может быть избирательно направлены на цели (или активированы или деактивированы) и они могут далее быть адаптированы к моделям прогрессирующих болезней. Фактически, исследования in vivo, использующие разные модели болезней уже начаты, чтобы пролить свет на относительный вклад отобранных рецепторных путей для терапевтического использования. Некоторые успехи в этом направлении включают характеризацию β-arrestin 2-biased лигандов для типа 1 angiotensin II рецептора у мышиных моделей кардиомиопатии 156,157 и анализ in vivo обезболивающих и побочных эффектов профилей G белков, вызывающих склонность у κ-OR 158 и µ-OR1 59,160. Важно, что исследование систематически анализирующее коллекцию дифференциально biased лигандов для µ-OR недавно продемонстрировало положительную корреляцию между их степенью, обеспечиваемой с помощью G белков передачи сигналов, над рекрутированием arrestin (G protein bias), и шириной их терапевтического окна (баланс между терапевтической эффективностью и побочными эффектами), предоставляя основы для разработки терапевтических средств с улучшенным клиническим эффектом для этого класса лекарств 161. На уровне обнаружения лекарств мы можем теперь использовать наши знания о структуре и динамике рецепторов, возможности химической модификации этого типа тенденциозных передач сигналов и разнообразить лекарственные реакции, чтобы использовать multiplexed и кинетические измерения клеточных функций для улучшения фармакологического кластрирования химиотипов (Box 1). Самостоятельные фармакологические химиотипы могут быть затем эмпирически связаны с желательными эффектами in vivo.
Understanding the impact of disease context on drug action
Патологические состояния могут модифицировать клеточные функции, включая альтерации локального окружения рецепторов. Один пример изменения композиции мембраны, который может наблюдаться при метаболических болезнях, таких как гиперлипидемия, ожирение и диабет 162 и при нарушениях ЦНС, включая болезнь Алцгеймера и шизофрению 163. События, такие как гипоксия, изменения pH и альтерации redox потенциала и генерализованной воспалительной реакции, все они могут изменять локальное клеточное окружение и тем самым потенциал реакции на лекарства 164-168. Одним из примеров, где такие изменения происходят, является микроокружение опухоли, которое часто кислое. Очевидно, GPCRs содержат сети остатков. которые могут быть присоединять протоны и являются чувствительными к изменению pH, при этом альтерации этих сетей могут приводить к последствиям активации рецепторов. В частности, некоторые GPCRs, как полагают, функционируют как сенсоры протонов путем альтераций состояний protonation законсервированных негативно заряженных остатков (таких как D3.32 и D2.50 (using the class A GPCR numbering scheme 68)) , также как и заряд погруженных гистидинов 168,169, это может приводить к изменению передачи сигналов рецепторами при низком pH окружении. Альтерации в уровнях др. ионов также были рассмотрены, когда оценивали передачу сигналов GPCR. Важно. что катионы, такие как Na+, находятся среди наиболее распространенных аллостерических модуляторов функции GPCR и вносят вклад в активность рецепторов (молчание в противовес конституитивной активации) и в природу тенденциозной передачи сигналов 69,170,171. Т.к. такая природа реакций GPCR на лекарства может быть изменена драматически при разных контекстах болезни, приводя, напр., к различиям в использовании трансдуцеров и регуляторных белков (т.е., изменяя наблюдаемую тенденциозность профиля лигандов). Более того, большинство болезней являются гетерогенными и используют это по мере прогрессирования. Следовательно, использование лекарственного вмешательства, оптимального для данной ст. болезни, является важным компонентом для поступательного успеха. Более сложные модельные системы, воспроизводящие контекст болезни являются важными для понимания действенности в поступательном движении и для становления степени, с которой кластеризуется лекарственное поведение в рутинных рекомбинантных клеточных исследованиях остается прогнозируемым для меняющихся условий.
Considering the impact of natural receptor variation on GPCR-drug interaction
Др. ключевым соображением, которое может влиять на обнаружение лекарств, разработку и в конечном итоге на успешное использование, является генетическая изменчивость последовательностей рецепторов (полиморфизмы) и изменчивость в экспрессии рецепторов между индивидами. Несколько независимых исследований охарактеризовали функциональное влияние, включая отличающиеся реакции на лекарства, количество полиморфизмов в коллекции рецепторных типов 172-174. Недавно проанализированы геномные последовательности у 68496 индивидов, показавшие, что на сегодня GPCRs обнаруживают значительную генетическую изменчивость внутри функциональных регионов, таких как связывающих лекарства и места, связывающие трансдуцеры 19. Экспериментальное исследование µ-OR выявило, что индивиды с определенными полиморфизмами, обладают неожиданными реакциями на агонисты, частичные агонисты и антагонисты. Напр., полиморфизмы в позиции вблизи кармана, связывающего лекарство, обнаруживают избыточный функциональный эффект, при этом антагонист или частичный агонист могут вызывать полную реакцию, характерную ля агониста. Исследования cholecystokinin receptor type A далее подтвердили, что определенные полиморфизмы вблизи региона, связывающего G белок этого рецептора полностью меняют профиль селективности к связыванию трансдуцера 19. Сходным образом, генетическая изменчивость может менять использование рецептора с arrestins, чтобы повлиять на регуляцию рецептора и/или нижестоящей передачи сигналов для CXC-chemokine receptor 4 (ref.175) и vasopressin рецепторов 176. Такая измененная или избыточная функция может вызывать тяжелые побочные эффекты на лекарства и возможно угрожать жизни. Эти находки подчеркивают необходимость понимания и характеристики вариантов рецепторов, которые превалируют в популяции человека и учитывать их при обнаружении лекарств. Характеризация вариантов GPCR как мишеней для лекарств также может помочь в уточнении предписания лекарства пациентам из определенных генотипических групп с предсказуемыми реакциями на лекарства. Такая стратификация д. повысить надежность и эффективность лекарств и как следствие д. улучшить качество жизни пациентов, экономические и социальные затраты.
Conclusions and perspectives
Biased agonism is a fundamental property of GPCR ligands that can be used - serendipitously or constructively - for improved therapeutic targeting of these receptors. Consideration of this aspect of GPCR pharmacology will be key to achieving clinical drug efficacy and safety. Our understanding of drug action is limited to what we measure as a drug response, and high-resolution structural insights into the control of GPCR conformation are still at an early stage, providing limitations to conscious application of the principle of biased agonism in drug design. Advances in structural approaches to understand drug-receptor interaction and the increasing ability of researchers to analyse multiple signalling end points and to measure these events in real-time are progressively enabling integration of a breadth of cellular signalling outcomes, and of the kinetic and spatial elements of GPCR signalling, into structure-based drug discovery. This progress is expected to improve drug candidate selection. Nonetheless, applying the biased agonist properties of ligands for improvement of the therapeutic effect of new drugs will require an understanding of the expression of receptors and components of their signalling pathways in different tissues and of the tissue-specific and context-specific, integrated signalling that differentiates beneficial from detrimental effects. Given this complexity, rational application of biased agonism is expected to have the greatest impact on the treatment of diseases for which pathological mechanisms are well characterized177.
|