Посещений: 
ПРОСТРАНСТВЕННАЯ ПРОТЕОМИКА
Подходы к изучению
Spatial proteomics: a powerful discovery tool for cell biology • Emma Lundberg &
• Georg H. H. Borner
 Nature Reviews Molecular Cell Biologyvolume 20, pages285–302 (2019)
| |
|
Клетки эукариот сильно компартментализованы, чтобы обеспечить разграничение биологических процессов. Компатменты включают разнообразные связанные с мембранами органеллы, такие как аппарат Гольджи, endoplasmic reticulum (ER) и митохондрии, а также лишенные мембран структуры, такие как крупные белковые ансамбли (напр., центросомы и фокальные адгезии) и разделенные по фазе капли (напр., РНК гранулы1,2). Функции белков тесно связаны с субклеточной локализацией, т.к. разные компартменты предоставляют разное химическое окружение (такое как pH и состояние redox), потенциальных партнеров по взаимодействию или субстраты. Тонкая регуляция субкл6еточной локализации белков поэтому является важной для контроля клеточной физиологии3. Большинство клеточных биологических процессов используют изменения в субклеточной локализации белков, такие как снование между ядром и цитозолем транскрипционных факторов, перемещения митохондриальных белков во время апоптоза и endocytic потребления грузов рецепторами клеточной поверхности и сигнальными рецепторами. Напротив, неправильная локализация белков часто ассоциирует с дисфункцией клеток и болезнями, включая не1йродегенерацию, рак и метаболические нарушения4-7. Знание пространственного распределения белков на субклеточном уровне и способность определять динамику субклеточных белков поэтому важны для полного понимания клеточной биологии.
Недавние успехи в высокопроизводительной микроскопии8, в quantitative mass spectrometry (MS)9-12 и в межатомном картировании13,14, а также в обучающихся машинных приложениях для анализа данных15-17, сделали возможными протеомные исследования пространственной клеточной регуляции. Обычно клетки человека экспрессируют более 10000 разных белков, охватывающих диапазон численности семи порядков18. Современные крупномасштабные исследования пространственного протеома человека указывают на то, что существует довольно сложная архитектура, которая включает изменчивость между одиночными клетками (на уровне и локализации белка), динамической транслокации белков, изменений в сети взаимодействий и локализации примерно половины из всех белков по многим компартментам (подразумевая потенциальную активность "подрабатывать"('moonlighting' )). Включение всего количества данных позволяет построить клеточную модель, а системный анализ неоходим после количественного описания19-23. Более того, некоторые исследования успешно использовали силу глобальной пространственной протеомики для изучения болезней, включая острую вирусную инфекцию24 и печеночные болезни25 или точечных дефектов в клетке, которые лежат в основе моногенных нарушений26,27.
Доступные на сегодня подходы к пространственной протеомике хорошо дополняют др. др. и их индивидуальные преимущества и ограничения делают их пригодными для разного типа приложений. Пространственная протеомика обладает значительным потенциалом, чтобы принести пользу во многих областях клеточной биологии и становится технически доступной.
В этом обзоре мы используем термин пространственная протеомика, чтобы обозначить специфически субклеточное картирование белков; примеры многочисленных макроскопических протеомных картирований органов и тканей мы не будем обсуждать, включая атлас тканей человека 28, атлась головного мозга мыши 29 и атлас сердца человека 30
.
Spatial proteomics methods
В целом три комплементарных подхода используются в пространственной протеомике: MS анализ фракционированных органелл, анализ сетей межбелковых взаимодействий и протеомное изображение локализации белков (Figs 1,2,3; Supplementary Table 1).
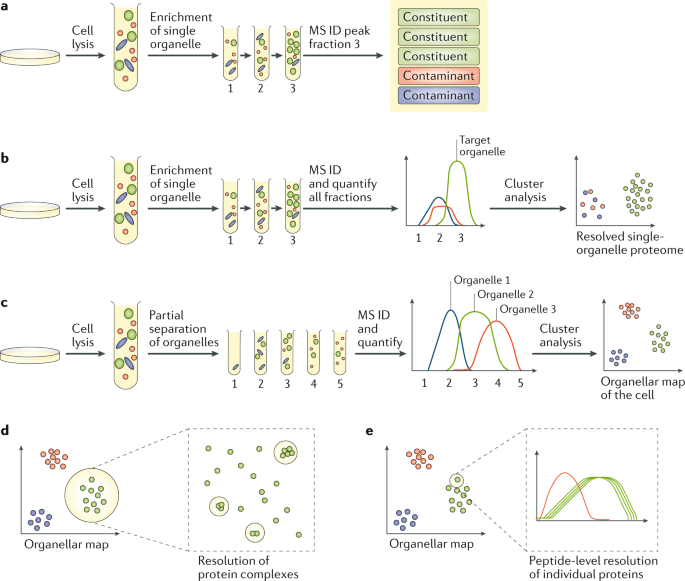 Fig. 1: Spatial proteomics by MS analysis of fractionated organelles.
a | In conventional organelle enrichment, cells are lysed, and tailored subcellular fractionation (for example, gradient centrifugation or differential centrifugation) is carried out to enrich a target organelle (green), followed by analysis with mass spectrometry (MS), of the most enriched fraction only. Identified proteins include genuine constituents of the target organelle and co-enriching contaminants (red and blue), which cannot be objectively distinguished; consequently, this approach is not recommended. b | In single-organelle profiling, cells are lysed, and tailored subcellular fractionation is carried out (as in part a) to enrich a target organelle (green), followed by quantitative MS of the enriched fraction and one or more of the subfractions from the enrichment protocol (such as neighbouring fractions on a gradient or crude fractions). For each protein, an abundance distribution profile is obtained. Proteins associated with the target organelle (green) have similar profiles and can be discriminated from contaminants (red and blue) by statistical analysis. Contaminants are recognized as such but are not necessarily resolved into distinct classes. c| In multi-organelle profiling, cells are lysed, and a subfractionation protocol is applied to partially separate all organelles simultaneously (only three are shown to illustrate the principle). No organellar 'purification', as such, is attempted; organelles have largely overlapping distribution. Quantitative MS is then carried out on the subcellular fractions. Each organelle has its own distinct profile, which is shared by all proteins predominantly associated with this organelle. Protein profiles are deconvolved by cluster analysis, and annotation of the plot with established organelle markers reveals cluster identities. In the obtained organellar map, the profile of each protein is represented by a dot; the position indicates the organellar association of the protein. d | Organellar maps have high local resolution. Proteins that are part of the same complex have tightly linked fractionation profiles that appear as microclusters within organellar maps, a feature that can be used to identify (ID) novel protein complexes40. e | Organellar maps can provide peptide-level resolution. Quantification by MS works at the level of peptides. The fractionation profile of a protein (a dot on the organellar map, left panel) is an average (or median) of the profiles from all peptides matching the sequence of that protein. Peptide profiles from the same protein tend to be closely aligned (right panel, green). However, if a post-translational modification (such as phosphorylation) causes a shift in the subcellular localization of a protein, the peptide containing the modification will have a different profile (red). As usually only a fraction of the copies of a protein present within a cell are modified, the profile of the modified peptide is different from that of the bulk of the other peptides.
Full size image
Fig. 1: Spatial proteomics by MS analysis of fractionated organelles.
a | In conventional organelle enrichment, cells are lysed, and tailored subcellular fractionation (for example, gradient centrifugation or differential centrifugation) is carried out to enrich a target organelle (green), followed by analysis with mass spectrometry (MS), of the most enriched fraction only. Identified proteins include genuine constituents of the target organelle and co-enriching contaminants (red and blue), which cannot be objectively distinguished; consequently, this approach is not recommended. b | In single-organelle profiling, cells are lysed, and tailored subcellular fractionation is carried out (as in part a) to enrich a target organelle (green), followed by quantitative MS of the enriched fraction and one or more of the subfractions from the enrichment protocol (such as neighbouring fractions on a gradient or crude fractions). For each protein, an abundance distribution profile is obtained. Proteins associated with the target organelle (green) have similar profiles and can be discriminated from contaminants (red and blue) by statistical analysis. Contaminants are recognized as such but are not necessarily resolved into distinct classes. c| In multi-organelle profiling, cells are lysed, and a subfractionation protocol is applied to partially separate all organelles simultaneously (only three are shown to illustrate the principle). No organellar 'purification', as such, is attempted; organelles have largely overlapping distribution. Quantitative MS is then carried out on the subcellular fractions. Each organelle has its own distinct profile, which is shared by all proteins predominantly associated with this organelle. Protein profiles are deconvolved by cluster analysis, and annotation of the plot with established organelle markers reveals cluster identities. In the obtained organellar map, the profile of each protein is represented by a dot; the position indicates the organellar association of the protein. d | Organellar maps have high local resolution. Proteins that are part of the same complex have tightly linked fractionation profiles that appear as microclusters within organellar maps, a feature that can be used to identify (ID) novel protein complexes40. e | Organellar maps can provide peptide-level resolution. Quantification by MS works at the level of peptides. The fractionation profile of a protein (a dot on the organellar map, left panel) is an average (or median) of the profiles from all peptides matching the sequence of that protein. Peptide profiles from the same protein tend to be closely aligned (right panel, green). However, if a post-translational modification (such as phosphorylation) causes a shift in the subcellular localization of a protein, the peptide containing the modification will have a different profile (red). As usually only a fraction of the copies of a protein present within a cell are modified, the profile of the modified peptide is different from that of the bulk of the other peptides.
Full size image
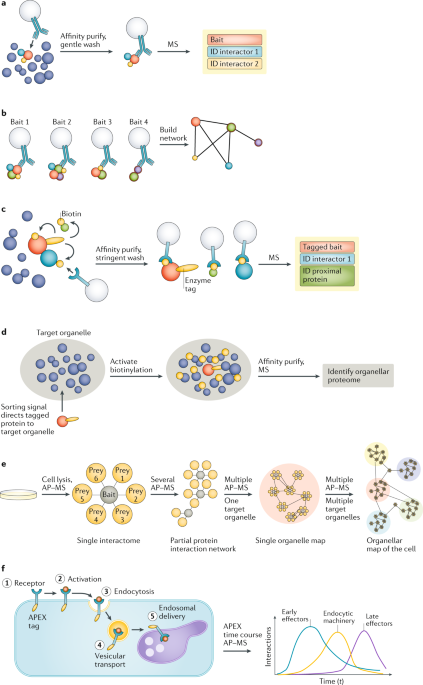 Fig. 2: Spatial proteomics through interaction networks.
a | Affinity purification-mass spectrometry (AP-MS). Proteins and their binding partners can be affinity purified from complex mixtures, such as whole-cell lysates, using antibodies. Gentle washing conditions are required to preserve protein-protein interactions, and MS analysis is used to identify (ID) the recovered proteins. b | AP-MS networks. The interactome of a single protein is a 'local' spatial proteome, as interacting proteins must be in the same subcellular location. Carrying out multiple AP-MS experiments with interacting baits reveals a network of associations that also has spatial information. c | Proximity labelling. A bait protein can be tagged with an enzyme (such as engineered ascorbate peroxidase (APEX) or a promiscuous biotin ligase in the biotin identification (BioID) method) that catalyses the biotinylation of proteins in close proximity (<10-20?nm). Targets usually include the tagged protein itself and direct binding partners but can also include transient interactors or even closely juxtaposed (but not directly binding) proteins. As the modification is covalent, biotinylated proteins can be recovered using affinity purification under stringent conditions before identification by MS. d | Spatial proteomics through proximity labelling. Proteins tagged with APEX or a promiscuous biotin ligase (BioID) can be chosen or engineered to target a specific organelle (as illustrated here) or subcellular location (such as the synapse). Activation of the enzyme causes widespread biotinylation of nearby proteins, which can be identified using MS. Thus, comprehensive compartment proteomes can be obtained without the need for subcellular fractionation. e | Spatial proteomics through expanding interaction networks. When multiple AP-MS or proximity labelling experiments are carried out in the same system, the baits and binding partners begin to overlap (as in part b). The use of multiple baits from the same subcellular localization yields detailed maps of individual compartments. Mapping of multiple compartments reveals connections between them, ultimately providing a spatial map of the cell. f | Spatiotemporal mapping of protein associations. Proximity labelling can be used to reveal the changing interactions of signalling receptors during their activation (by ligand binding), endocytic uptake and vesicular delivery to the endosomal system (left side of schematic) at subminute temporal resolution. The fluctuating abundance of groups of interacting proteins identified at different time points is illustrated (right side).
Fig. 2: Spatial proteomics through interaction networks.
a | Affinity purification-mass spectrometry (AP-MS). Proteins and their binding partners can be affinity purified from complex mixtures, such as whole-cell lysates, using antibodies. Gentle washing conditions are required to preserve protein-protein interactions, and MS analysis is used to identify (ID) the recovered proteins. b | AP-MS networks. The interactome of a single protein is a 'local' spatial proteome, as interacting proteins must be in the same subcellular location. Carrying out multiple AP-MS experiments with interacting baits reveals a network of associations that also has spatial information. c | Proximity labelling. A bait protein can be tagged with an enzyme (such as engineered ascorbate peroxidase (APEX) or a promiscuous biotin ligase in the biotin identification (BioID) method) that catalyses the biotinylation of proteins in close proximity (<10-20?nm). Targets usually include the tagged protein itself and direct binding partners but can also include transient interactors or even closely juxtaposed (but not directly binding) proteins. As the modification is covalent, biotinylated proteins can be recovered using affinity purification under stringent conditions before identification by MS. d | Spatial proteomics through proximity labelling. Proteins tagged with APEX or a promiscuous biotin ligase (BioID) can be chosen or engineered to target a specific organelle (as illustrated here) or subcellular location (such as the synapse). Activation of the enzyme causes widespread biotinylation of nearby proteins, which can be identified using MS. Thus, comprehensive compartment proteomes can be obtained without the need for subcellular fractionation. e | Spatial proteomics through expanding interaction networks. When multiple AP-MS or proximity labelling experiments are carried out in the same system, the baits and binding partners begin to overlap (as in part b). The use of multiple baits from the same subcellular localization yields detailed maps of individual compartments. Mapping of multiple compartments reveals connections between them, ultimately providing a spatial map of the cell. f | Spatiotemporal mapping of protein associations. Proximity labelling can be used to reveal the changing interactions of signalling receptors during their activation (by ligand binding), endocytic uptake and vesicular delivery to the endosomal system (left side of schematic) at subminute temporal resolution. The fluctuating abundance of groups of interacting proteins identified at different time points is illustrated (right side).
 Fig. 3: Different approaches to imaging-based spatial proteomics.
a | Antibody-based visualization. Endogenous proteins are visualized using antibodies and immunofluorescence after initial fixation and permeabilization of the cells. For large-scale spatial proteomics studies, the assays are carried out on a massive scale in parallel using robotic pipetting devices. Images of the immunostained cells are acquired using high-throughput fluorescence microscopy. b | Fluorescent protein (FP)-tagging-based visualization. Gene-editing technologies are used to tag proteins with an FP reporter, which enables visualization of the protein's localization. Images of live cells are acquired using high-throughput fluorescence microscopy, either in time-lapse series or at static end points. c | Cellular organelles and structures. Example images of 12 cellular organelles and structures obtained using antibody-based visualization and fluorescence microscopy are shown. The marker protein for the structure of interest (green) is visualized together with markers for microtubules (red) and the nucleus (blue) to facilitate pattern recognition. d | High spatial resolution. Images demonstrate the great spatial detail obtained with confocal microscopy, which can pinpoint protein localization to different substructures of the cytokinetic bridge during cell division. CDCA8 is localized to the entire cytokinetic bridge, whereas APC2 is restricted to the midbody, and CBLN4 is restricted to the midbody ring. e | Multilocalizing proteins. HER2 is localized to both the plasma membrane (**) and the nucleus (*) in the same cell. In addition to its canonical function as a receptor tyrosine kinase at the plasma membrane, HER2 has been shown to 'moonlight' as a transcription cofactor in the nucleus. f | Single-cell variability. Enolase 1 is a cytosolic metabolic enzyme that moonlights as a DNA-binding protein in the nucleus; single cells with only cytoplasmic localization (*), only nuclear localization (**) or nuclear and cytoplasmic localization (***) are observed. The level of expression (that is, the staining intensity) is also highly variable between cells. Scale bar is 20 ?m. ER, endoplasmic reticulum. The images in parts c-f are from the Cell Atlas52 of the Human Protein Atlas.
Fig. 3: Different approaches to imaging-based spatial proteomics.
a | Antibody-based visualization. Endogenous proteins are visualized using antibodies and immunofluorescence after initial fixation and permeabilization of the cells. For large-scale spatial proteomics studies, the assays are carried out on a massive scale in parallel using robotic pipetting devices. Images of the immunostained cells are acquired using high-throughput fluorescence microscopy. b | Fluorescent protein (FP)-tagging-based visualization. Gene-editing technologies are used to tag proteins with an FP reporter, which enables visualization of the protein's localization. Images of live cells are acquired using high-throughput fluorescence microscopy, either in time-lapse series or at static end points. c | Cellular organelles and structures. Example images of 12 cellular organelles and structures obtained using antibody-based visualization and fluorescence microscopy are shown. The marker protein for the structure of interest (green) is visualized together with markers for microtubules (red) and the nucleus (blue) to facilitate pattern recognition. d | High spatial resolution. Images demonstrate the great spatial detail obtained with confocal microscopy, which can pinpoint protein localization to different substructures of the cytokinetic bridge during cell division. CDCA8 is localized to the entire cytokinetic bridge, whereas APC2 is restricted to the midbody, and CBLN4 is restricted to the midbody ring. e | Multilocalizing proteins. HER2 is localized to both the plasma membrane (**) and the nucleus (*) in the same cell. In addition to its canonical function as a receptor tyrosine kinase at the plasma membrane, HER2 has been shown to 'moonlight' as a transcription cofactor in the nucleus. f | Single-cell variability. Enolase 1 is a cytosolic metabolic enzyme that moonlights as a DNA-binding protein in the nucleus; single cells with only cytoplasmic localization (*), only nuclear localization (**) or nuclear and cytoplasmic localization (***) are observed. The level of expression (that is, the staining intensity) is also highly variable between cells. Scale bar is 20 ?m. ER, endoplasmic reticulum. The images in parts c-f are from the Cell Atlas52 of the Human Protein Atlas.
MS-based organelle profiling
MS может быть использована для идентификации белков и их численности в очень сложных смесях 9-11. Традиционный биохимический подход к пространственной протеомике используется, чтобы обогатить определенными органеллами с помощью субклеточного фракционирования, что сопровождается идентификацией белков в самой многочисленной фракции с помощью MS (Fig. 1a). Для гомогенных компартментов с характерными размерами, плотностью и формой, может быть достигнуто существенное обогащение (напр., синаптических пузырьков 31). Однако, хотя подход по выделению органелл всё ещё широко распространен 32, теперь считается, что большинство субклеточных компартментов делают невозможной полную 'очистку', из-за их прирожденной гетерогенности и перекрывания физических свойств. Т.о., детекция белка во фракции обогащенных определенных органелл является недостаточным доказательством их специфической ассоциации с органеллами. Появление количественной MS предоставило значительный прогресс в этом отношении, а соотв. экспериментальные настройки могут помочь отличать между составными частями компартментов и неспецифическим фоном беспристрастным способом. Подходы по профилированию этих органелл были применены к одиночным органеллам и, что важно, к целым клеткам, дали всеобъемлющие высокого разрешения карты органелл клеток.
Single-organelle profiling
Основной стратегией профилирования органелл является завершение специального фракционирования для обогащения специфическими органеллами и затем количественная оценка белков на разных ступенях протокола обогащения, используя MS (Fig. 1b). Для каждого белка получали профиль распределения. Белки, ассоциированные органеллами мишенями или компартментом, имели сходные профили и могут быть отличены от загрязнений, имеющих др. профили. Этот мощный подход 33 впервые был использован с MS при protein correlation profiling (PCP) центросом, очищенных в градиенте сахарозы 34 и стех пор успешно используется для определение протеомов некоторых органелл (включая капельки липидов 35, митохондрии 36,37, лизосомы 38 и транспортные пузырьки 39,40). Концептуально родственные исследования использовали похожие свойства для выделения интактных ядер из яиц Xenopus laevis с целью количественной оценки белков в ядрах и цитозоле, чтобы выяснить аккуратно распределения межу ядром и цитозолем 41 и профилирования плазматических мембран в сравнении с протеомом целой клетки после инфекции human cytomegalovirus (HCMV) в фибробластах, чтобы понять, как вирус преобразует поверхность клетки, чтобы преодолеть иммунную защиту 42. Более того, профилирование комбинировали с устранением или модификацией компартмента. Т.о., анализ фракции, обогащенной митохондриями перед и после истощения аппарата митохондриального импорта позволил идентифицировать митохондриальный 'importome' у трипаносом; этот подход оказался также успешным для характеристики пероксисом 43. Сходным образом, профилирование фракций, обогащенных пузырьками, которые были истощены по разным ключевым белкам из аппарата транспорта clathrin выявило состав разного типа покрытых клатрином пузырьков 39. Все эти исследования достигли высокого уровня чувствительности и специфичности.
Multi-organelle profiling to create MS-based maps of the cell
Концепция профилирования одного типа органелл может быть расширена на все субклеточные компартменты одновременно. Чтобы достичь этого клетки лизируют механически, чтобы высвободить (в идеале нетронутые) органеллы, которые затем частично разделяют с помощью градиентного центрифугирования (Fig. 1c). Хотя пик разных органелл приходится на разные фракции, они обычно обнаруживают существенное перекрывание распределения. Затем фракции из градиента анализируются с помощью количественной MS , чтобы определить профиль количественного распределения для каждого белка. Кластерный анализ выявил группы белков со сходными профилями, которые соответствовали разным органеллам. На кластеры затем накладывали известные маркеры органелл, чтобы выявить принадлежность кластера. Наконец, использовали профили маркеров, чтобы получить алгоритм классификации (такой как support vector machines или нейральные сети) для предсказания ассоциации компартмента из немаркированных белков. Это давало детальную карту органелл клеток, указывающую на субклеточную локализацию определяемых количественно белков.
Осуществление профилирования многих органелл достигло заметного улучшения разрешения и применимо к большинству субклеточных компартментов в одном эксперименте и стало инструментом анализа на системном уровне (see below and Box 1). Экспериментально профилирование множественных органелл также сравнительно быстрое и технически несложное (Supplementary Box 1). Напротив, косвенные взаимодействия субклеточных локализаций не всегда очевидны, т.к. профили отражают среднее устойчивое состояние распределения белков. Профили белков, которые преимущественно ассоциируют с одним типом органелл, легко интерпретируются путем сравнения с профилями маркеров органелл. Однако, для белков. находящихся во многих компартментах, получаются смешанные профили, это приводит к неопределенности классификаций или даже к ошибочным классификациям44. Анализ смешанных профилей является одной из величайших проблем в подходах профилирования, особенно потому, что почти нет количественных эталонных данных по multi-organellar распределению. Как результат большинство экспериментов по multi-organelle профилированию использует MS, включая относительно высокую пропорцию (обычно 25-45%) белков, которые не могут быть с уверенностью приписаны компартменту. Др. ограничением является то, что только компартменты, для которых существуют реальные маркеры, могут быть включены в схему классификации45, хотя de novo открытие компартментов является возможным в принципе46. Более того, профилирование органелл 'усредняет' распределение белка от миллионов клеток, которые используются для биохимического фракционирования, приводя к потере определенной информации о межклеточной изменчивости.
Некоторая изменчивость при multi-organelle профилировании была определена20,21,25,47-51, которая отличалась от таковой при использовании метода отдельных органелл, при MS количественной стратегии и биоинформационном анализе. Три подхода испоьзуются во многих экспериментальных контекстах: а именно, PCP25, локализация белков органелл с помощью мечения изотопами (LOPIT)48 и динамическое картирование органелл21 (see Box 1 for a detailed comparison of these approaches).
Независимо от использованного метода, все подходы multi-organelle профилирования, опубликованные в последни 3 года20,21,25,26,47,52 достигли очень высоких уровней разрешения органелл (~10 субклеточных компартментов), протеомного покрытия (более 5000 белков) и аккуратности классификации (k.sxuk оценивается более 90%). Предсказания по компартментной локализации, полученные с использованием LOPIT и динамического картирования органелл также были сравненны непосредственно21; совпадение было подсчитано выше 91%, это строго подтверждает аргументированность результатов, полученных любым из этих методов. Эти исследования предоставили детальный состав (inventories) органелл клеток печени мыши25, нейронов20 и эмбриональных стволовых клеток47, а также клеток печени крыс50, клеток HeLa 21 и клеток USO252, сделав возможным впервые систематические сравнения пространственных протеомов у разных типов клеток и видов20,52.
Благодаря высокой точности современной MS количественной оценки, профили индивидуальных субъединиц из стабильных мультимерных белковых комплексов часто настолько сходны с теми, которые были идентифицированы как плотные микрокластеры внутри карт органелл21,47 (Fig. 1d; discussed further elsewhere40). Т.о., карты органелл в действительности предоставляют большое количество interactomics данных, которые должны все же систематически добываться. Более того, белковые профили сами по себе являются средними от множественных индивидуальных пептидов. Карты органелл могут т.о., в принципе, предоставлять разрешения на уровне пептидов (Fig. 1e) и выявлять различия в локализации сплайс-изоформ белков и протеолитически преобразованных форм, также как и различия в локализации, связанные с пост-трансляционными модификациями. Напр., фосфорилированные пептиды и их немодифицированные аналоги четко различаются с помощью MS. Если субклеточная локализация белков изменяется в ответ на фосфорилирование (как в случае большинства транскрипционных факторов), тогда модифицированный и не модифицированный пулы пептидов имеют разные субклеточные локализации, что доказывается их разными положениями на картах органелл на уровне пептидов. Это удивительное свойство только начинает эксплуатироваться, поскольку это напрягает существующую технологию до крайних пределов, при этом относительно мало демонстраций, доказывающих принцип (напр., для сплайс-форм47 и по-разному расположенные фосфопептидов25). Тем не менее мы ожидаем, что картирование органелл на уровне пептидов вскоре внесет существенный вклад в раскрытие субклеточной локализации сложных белковых форм.
|
Box 1 Current implementations of MS-based multi-organelle profiling
Three major methods of mass spectrometry (MS)-based multi-organelle profiling are currently used, namely, protein correlation profiling (PCP)142, localization of organelle proteins by isotope tagging (LOPIT)48 and dynamic organellar maps21, which all offer comparable levels of compartment resolution and proteome coverage. Although the methods are conceptually similar, differences in organelle separation and MS quantification strategies result in individual-specific and application-specific advantages (see the table for a comparison of these MS-based multi-organelle profiling methods). Of note, all three methods have been applied successfully to generate extensive databases of protein subcellular localization (see below) and have also been used in comparative studies as discovery tools (see Global comparative spatial proteomics). |
Protein correlation profiling
Органеллы разделяются с помощью скоростного центрифугирования в градиенте плотности сахарозы и ~20 субфракций было проанализировано с помощью лишенной метки количественной оценки MS. Самое большое количество субфракций, которые могут быть количественно разделены в PCP до определенной степени, выявляют мультимодальные распределения органелл, делая тем самым возможным распределение белков по многим субклеточным локализациям, это является ключевой основой этого подхода. PCP был применен как к дикого типа, так и к steatotic мышиным клеткам печени и это предоставило информацию о расположении более 6000 белков, которые были картированы в десяти субклеточных компартментах 25 (см. базу данных obesity-induced non-alcoholic fatty liver disease (NAFLD)).
Localization of organelle proteins by isotope tagging
Органеллы, разделяемые центрифугированием в градиенте плотности, и десятки фракций отбираются для дальнейшего анализа и количественной оценки с использованием tandem mass tag multiplexing (TMT multiplexing). LOPIT предоставляет хорошее разрешение для органелл; впервые разработан для определения эндомембранной системы у растений 141, усовершенствованная версия (hyper-LOPIT) была применена к стволовым клеткам мышей, обеспечив в результате профилирование более 5000 белков и картирование 14 компартментов 47(see the SpatialMap database). Более того, LOPIT картирование U2OS клеток впервые сделало возможным прямое крупномасштабное сравнение данных по MS-based и imaging-derived локализации белков 52, которые обнаруживали высокий уровень сходства классификаций между этими ортогональными подходами.
Dynamic organellar maps
Органеллы были разделены с помощью дифференциального центрифугирования на 8 фракций. Параллельно, общая фракция мембран была приготовлена из метаболически меченных клеток и эта фракция добавлялась к каждой фракции в качестве внутренней оценки для квантификации перед MS анализом. Этот мощный производственный процесс давал высоко воспроизводимые карты, делая возможным тем самым чувствительный сравнительный анализ (т.е до и после пертурбаций). Метод был применен к клеткам HeLa, и сделал возможным картирование 8700 белков в 15 компартментах 21 (see the HeLa Spatial Proteome database). Конечно трудовой процесс был также исполнен с отсутствием метки количественным процессом и TMT multiplexing; использование лишенного метки формата сделало возможным детальное картирование органелл для первичных нейронов мыши (8000 белков и 12 компартментов 20). Более того, динамическое картирование органелл зарегистрировало абсолютные количества белка (т.е. количества копий белка на клетку), так что были получены количественные модели органелл и клеточного состава 20,21.
Table 1
Spatial protein interaction networks
MS активно использовали, чтобы определять межбелковые взаимодействия, используя эксперименты по антителами обусловленной очистке-MS (AP-MS) (Fig. 2a). Концептуально интерактом белков является 'локальным' пространственным протеомом, т.к. белки д. занимать одни и те же места, чтобы взаимодействовать. После анализа достаточно большого количества наживки из одной и той же системы, картированные взаимодействия в конечном итоге формировали взаимосвязанную сеть, которая предоставляла информацию о субклеточной локализации белков (Fig. 2b). Два наиболее обширных крупномасштабных interactomics скринингов в клетках млекопитающих были скомбинированы, включали несколько тысяч baits53,54; однако, полного покрытия протеома и систематической пространственной интерпретации взаимодействий всё же не достигли. Несмотря на это подход быд успешно использован для картирования протеомов одиночных органелл. Базовой стратегией стала идентификация партнеров по взаимодействию некоторых белков, которые, как известно, располагаются в органеллах мишенях. Т.к. общая сложность протеомов органелл обычно составляет 1-2 порядка величин, меньшего, чем для клетки в целом20,21,47, то всесторонняя сеть ассоциаций может быть получена даже с небольшими наживками (baits). Прекрасным примером является выяснение композиции algal pyrenoid, лишенного мембран компартмента в хлоропластах, который чрезвычайно важен для концентрации и фиксации углекислого газа. Оказалось, что AP-MS осуществляется только с 38 baits55. Это исследование выявило множество деталей молекулярной архитектуры pyrenoid и проиллюстрировало также совметную деятельность, получаемую путем комбинирования AP-MS с получением ортогональных изображений.
Схожесть методов мечения особенно подходит для AP-MS картирования органелл и включения, помимо прочих, искуственно преоблазованой ascorbate peroxidase (APEX) и зависимой от близости biotin identification (BioID)13,14,56(Fig. 2c). В обоих этих методах, bait белков были нагружены энзимом, который biotinylates белки в тесной близи (обычно меньше10-20 nm), а меченные белки впоследствие обнаруживаются с использованием понижения рецептора streptavidin. Поскольку мечение не ограничивается прямыми партнерами по связыванию, то даже одиночная наживка (bait) может идентифицировать множество проксимальных белков, особенно в ограниченном пространстве органелл, предоставляя тем самым богатую пространственную информацию исходя из небольшого количества образцов (bait) (Fig. 2d). Оба метода были использованы для определения протеома органелл, дали превосходное покрытие и разрешение; примеры включают анализ митохондриальных субкомпартментов, используя APEX57,58, и разные типы РНК гранул, используя BioID59. Более того, энзимами нагруженные конструкции могут быть выбраны и преобразованы, чтобы их нацелить на очень специфические регионы клетки и тем самым предоставить информацию о компартментах, которые делают невозможным биохимическое выделение, таких как интерфейс центросома-ресничка60 и разные части синапса(revю 61).
В конечном итоге глубокое картирование индивидуальных органелл позволит идентифицировать связи между ними и теми, что начинают генерировать bottom-up, high-resolution, целой клетки пространственные протеомы (Fig. 2e). Прогресс в направлении этой цели сделан после генерации базы данных для органелл 'context' BioID протеомов из 18 субклеточных локализаций с использованием одного установленного маркерного белка для каждого из компартментов62. Эта база данных, хотя и не является исчерпывающей, уже может служить в качестве координатной сетки (reference grid) для предсказания субклеточной локализации белков на основании только AP-MS данных, исходя из разметок известных мест идентифицированных взаимодействий.
Зависимое от близости мечение также является мощным инструментом для картирования пространственно-временных взаимодействий. Проведены исследования 63,64 в комбинации с APEX proximity labelling с мониторингом высокого разрешения эндоцитоза G protein-coupled receptor (GPCR) (Fig. 2f). После активации путем связывания лиганда, GPCRs плазматической мембраны подвергаются быстрому эндцитозу с помощью clathrin-покрытых пузырьков и транспортируются в эндосомы. APEX мечение в разные промежутки времени после стимуляции позволяет картировать изменения interactome нагруженных GPCRs. Две методологические инновации стали ключевыми для успеха этих исследований: Во-первых, некоторые дополнительные APEX pulldowns с изучаемыми белками в разных компартментах вдоль маршрута эндоцитоза были включены, чтобы достичь высокой специфичности 63,и Во-вторых, протокол быстрого APEX-мечения был скомбинирован с multiplexed количественной MS , чтобы достичь меньше минуты временного разрешения 64.
Imaging-based spatial proteomics
Пространственная протеомика с использованием изображений предоставляет технологию для визуализации белков в их нативном клеточном окружении без необходимости лизиса клеток или физического выделения компартментов или органелл перед анализом протеома (Fig. 3). Эта локализация in situ белков делает детекцию белков с мультимодельным распределением по органеллам объяснимой. Фактически исследования по получению крупномасштабных изображений показывает, что многие белки располагаются во многих клеточных компартментах52,65. Более того, становится всё яснее, что популяции генетически идентичных клеток обнаруживают изменчивость в экспрессии и локализации белков52,66-68, напр., во время клеточной дифференцировки69,70, в ответ на средовые флюктуации67,69,71 и вследствие лечения лекарствами19,7-75. Этот феномен обозначается 'bet-hedging', он показывает, что существует прирожденная изменчивость внутри популяции клеток в ответ на стрессы индивидуальных клеток, которые служат в качестве стратегии рассредоточения риска, чтобы предоставлять долговременным популяциям преимущества в приспособляемости. Базирующиеся на изображениях технологии сами приводят к изучению такой изменчивости путем отлавливания пространственного распределения белка с разрешением в одну клетку.
Изображения требуют визуализации белков, обычно путем использования сродства к таким реагентам как антитела или путем экспрессии слияний флуоресцентных белков. Генерация антител и генетически модифицированы белков является затратной и требует времени, это накладывает ограничения применительно ко всему протеому. Однако, если подходящие реагенты и слияния с флуоресцентным белком достигнуты, то они предоставляют специфичные для белка возможности. которые могут быть использованы для pulldowns, это предоставляет дополнительные возможности для последующего анализа взаимодействий белков и комплексов.
Antibody-based protein visualization
Пространственная протеомика с помощью изображений присходит из визуализацими одиночных белков с помощью методов иммуно-флуоресценции76,77 (Fig. 3a). Преимущества такого типа анализа связаны с тем, что affinity реагенты способны делать видимыми продукты эндогенных генов и могут легко использоваться для изучения белков во многих разных типах клеток и выборках. Этот подход очень чувствителен, поскольку флуоресцентный сигнал может быть амплифицирован, напр., вторично антителами. Основной помехой в иммуно-флюоресценции является то, что внутриклеточное окрашивание требует фиксации и permeabilization клеток, это ограничивает использование статичными конечными измерениями. Более того, фиксация и permeabilization могут вносить артефакты в морфологию клеток и в худшем случае затрагивать расположение белка
78,79.
Исторически антитела наиболее широко используемые affinity реагенты, но успехи комбинаторной белковой химии позволили получить новые affinity реагенты из разных каркасов80. Независимо от типа affinity реагента, оценка его специфичности важна для избегания фальшивых результатов. Сравнительное исследование 500 белков, которые были локализованы с использованием флуоресцентных меток для белков и антител, показали, что перекрестная реактивность с ядерными компонентами является наиболее распространенным артефактом при локализации с помощью антител81. Знания об отсутствии надежных оценок и воспроизводимости исследований, базирующихся на антителах82 , привели к разработке указаний по валидация для конкретного приложения83 и пакетного цитирования84.
Из-за денежных и временных затрат возникла необходимость в генерации affinity реагентов для целых протеомов, количество опубликованных исследований глобальных пространственных протеомов всё ещё чрезвычайно мало. The Human Protein Atlas (HPA) оказался пионером в этой области последние 15 лет он имел целью картировать пространственное распределение всех белков во всех типах клеток тела человека и создать рессурс знаний для биологии человека28,52,85,86. По этой причине собрана коллекция антител почти всего протеома87,88 и проанализирована89-91. Частью этой работы было создание карт ссылок с подробным описанием внутриклеточного распределения протеома человека в большой панели линий клеток, которая была обозначена HPA Cell Atlas52 (Fig. 3c). Используя 14000 антител, было определено расположение 12003 белков с разрешением в одну клетку, из которых 5662 белка никогда не были экспериментально локализованы до этого. Пространственное распределение этих белков оказывалось в одной или нескольких структурах из 30 клеточных структур, используемых для комбинированного ручного или компьютерного15 анализа изображений. Высокого разрешения конфокальная микроскопия позволила локализовать белки в тонких структурах, таких как фибриллярный центр ядрышек, концы микротрубочек и субструктуры цитокиновых мостиков (Fig. 3d), а также палочки и кольца, которые являются функционально не охарактеризованными цитоплазматическими структурами. Неожиданно это исследование выявило, что более половины белков человека локализованы во многих компартментах. Впервые получены паттерны картирования с множественной локализацией (т.е., proteome connectivity) среди органелл в клетках (Fig. 3e). Более того, ~16% протеома обнаруживает изменчивость или на уровне экспрессии белка или на уровне пространственного распределения в одиночной клетке (Fig. 3f). Эти данные подчеркивают, что необходимы методы с разрешением для одиночной клетки и с multiplexed обнаружением, чтобы распутать ковариантные паттерны белков, а также временные характеристики для изучения динамики мультимодального распределения белка. Технологические разработки, такие как высоко multiplexed изображения, использующие нагруженные металлом антитела, и масс-цитометрию92-94 или ДНК штрих закодированные антитела и флуоресцентную микроскопию95,96, сделали возможным одновременный анализ более 50 белков с субклеточным разрешением, это проложило путь для анализа гетерогенности протеома органелл в контексте соседних клеток.
Visualization using fluorescent protein fusions
Инсерция генов, кодирующих флуоресцентные белки, в эндогенных геномных локусах делают возможными временные исследования динамики и локализации белков почти в эндогенном клеточном контексте (Fig. 3b). Это лучше всего иллюстрируется с помощью исследований почкующихся дрожжей Saccharomyces cerevisiae. Поскольку гомологичная рекомбинация очень эффективна у дрожжей, то кодирующая последовательность флуоресценного белка может быть легко внедрена в генетический локус с сохранением паттерна эндогенной экспрессии гена. Геномная коллекция GFP-нагруженных линий дрожжей делает возможной систематическую локализацию 75% от всех предсказанных белков S. cerevisiae по 22 разным категориям клеточных компартментов в условиях нормального роста в живых клетках65. Это исследование стало первым исследованием локализации белков на системном уровне у эукариот, которое в свое время предоставило информацию о локализации 1630 белков у дрожжей. Дальнейшее исследование слияния этих дрожжей с использованием флуоресцентной микроскопии установило пространственные карты дрожжевого протеома и предоставило независимые наборы данных о содержании белка19,67,73,74 (rev. elsewhere75). Затем были созданы дополнительные геномные данные дрожжевых библиотек; напр., подход SWAp-Tag был использован для создания нагруженной С-треминальной и N-терминальной коллекции97,98. Были сделаны и дальнейшие попытки выяснения организации протеома с высоким разрешением. Напр., точная локализация 200 белков в системе эндомембран дрожжей, используя ко-локализацию с 7 известными маркерами эндомембранных компартментов, это привело к идентификации новых грузов coatomer complex I (COPI)99. Благодаря этим исследованиям сегодня известны несколько дрожжевых imaging-based баз данных по локализации белков100-105.
Систематические исследования изображений с использованием флуоресцентно слитых белков также были проведены на клетках человека, хотя они в основном ограничены фракцией протеома. Пионерские исследования пространственной локализации белков человека были осуществлены с использованием эктопической экспрессии нагруженных кДНК. Субклеточная локализация 1600 была установлена с использованием временной трансфекции при открытой рамке считывания106. Сходным образом, аннотированная библиотека репортерных клонов была создана с помощью загрузки на экзоны с использованием ретровирусной доставки. Т.о., 2180 белков человека, слитых с YFP и экспрессирующихся со своих эндогенных промоторов, были локализованы с использованием time-lapse флуоресцентной микроскопии107-109. Затем разработка базирующихся на CRISPR-Cas9 методов преобразила нашу способность нагружать гены человека в их эндогенных локусах, путем облегчения гомологией управляемой репарации110,111 и позволила исследовать динамику субклеточной локализации кодируемых слитых белков под эндогенным регуляторным контролем112-115. Появляются стратегии по систематической загрузке у разных организмов и систем, таких как Drosophila melanogaster116 и разных типов клеток человека117,118, тогда как разработка self-complementing split флуоресцентных белков делает менее разрушительной загрузку меток119,120 и мечение больших количеств белков121. В целом разработка этих методов прокладывает путь к конструированию геномных эндогенно нагруженных библиотек белков в клетках людей.
Чувствительность всё ещё является ограничивающим фактором для приложений, использующих изображения флуоресцентных белков. Напр., было подсчитано, что только верхушка из 30% наиболее распространенных белков в клетках человека обнаруживается как GFP слитые белки с помощью прямой флуоресцентной микроскопии121. Чтобы смягчить эти ограничения, сигналы от белков с низкими частотами, могут быть усилены с использованием более ярких и более photostable флуоресцентных белков. Схемы мечения белков повторами от флуоресцентных белков также были разработаны121, хотя любое искусственное вмешательство в нативный белок создает риск вмешательства в его функцию, экспрессию и деградацию и локализацию. Таким образом, необходимо позаботиться о том, куда флуоресцентные белки будут вставлены. Кроме того, для оценки клеток, экспрессирующих меченные белки, необходимо убедиться, что обнаруживаемая локализация белка представляет эндогенную локализацию. Соответствие между флуоресцентными белками и подходами, базирующимися на антителах, в терминах расположения белков, составляет ~80%, с равными показателями локализации артефактов для двух методов; неправильная локализация эндомембранной системы наиболее распространенная проблема для слияний флуоресцентных белков81.
Хотя процедура геномного мечения трудоёмкая, значительные преимущества этого подхода позволяют полагать, что как только библиотека клеток будет завершена, сравнительные исследования локализации белка в живых клетках после различных возмущений можно будет легко проводить.
High-content microscopy
Независимо от того, как белки визиализируются, критической частью экспериментов по пространственной протеомике является получение изображений. Разработка автоматизированной флуоресцентной микроскопии была распространена за пределы изучения изображений до такой степени, что тысячи выборок могут быть теперь проанализированы при разных состояниях 8. Ключевыми соображениями для этих экспериментов являются следующие. Во-первых, необходимо для статических или динамических time-lapse изображений определить, какой метод визуализации наиболее подходит. Во-вторых, существует компромисс между разрешением и пропускной способностью. Низкое разрешение при более высокой скорости приобретения может быть предпочтительным при изучении живых клеток, напр., транслокации между цитозолем и ядром после пертурбаций, тогда как технологии высокого разрешения, такие как oil-immersion конфокальная микроскопия или super-resolution микроскопия (rev. elsewhere 122), могут выявлять чрезвычайно запутанные паттерны расположения на уровне суборганелл 22,52,123,124. В-третьих, клеточные контрольные отметки могут быть необходимы для сегментации клеток и ядер и для улучшения количества анализа изображений. Ядерные 74 и цитозольные 19,67 маркеры используются чаще всего, но маркеры эндомембранных систем 52,99 и цитоскелетные структуры 52 также должны быть включены для более утонченной оценки локализации.
Image analysis and location classification
Анализ набора данных изображений является др. критическим аспектом исследований пространственной протеомики. Др. ступень предварительной обработки, такая как нормализация и клеточная сегментация четко стандартизирована125, последующая ступень классификации может быть сильно затруднена. Целью этого анализа является распознавание субклетоного паттерна распределения белка и идельно также определение количества белков в каждом компартменте.
Распознавание паттерна вручную является преимущественным подходом; однако, для воспроизводимости, масштабируемости и быстроты анализа, необходимы автоматические решения (rev. elsewhere125). Попытки автоматичекой классификации базируются на методах, таких как K-nearest neighbour classifiers, support vector machines, artificial neural networks and decision trees, часто использующие набор свойств ловкости рук для определения белкового паттерна в соответствии с клеточной морфологией, включая от сотен до тысяч парамтров, описывающих форму, положение и текстуру окрашивания в связи с клеткой125-128. Сегодня глубокие нейральные сети16,129 приобрели популярность в качестве инструментов для анализа изображений130,131. Такие сетевые преобразования изображений посредством последующих слоёв вычисляемых единиц, которые количества всё возрастающих паттернов сложности в данных, и тренируют в предсказаниях данных меток в изображениях. Одной из сильных сторон этого подхода является то, что свойства изображений может быть получены автоматически. Однако, успешность глубоких нейральных сетей для анализа изображений критически зависят от доступности данных тренировок для изучения параметров моделей. Глубокие нейральные сети оказались успешными при классификации одномодально-локализованных белков у почкующихся дрожжей132,133 и в клетках человека133,134 пи этом аккуратность достигала 91%, но эти исследования оказались в основном ограниченными классификацией одиночных паттернов (9-18 locations) в одной клеточной линии. Последующие исследования были адресованы моделям, ручной классификации паттернов всё ещё со значительно высокой аккуратностью15. Прагматический среднего уровня подход использует беспристрастный компьютерный анализ изображений, чтобы идентифицировать интересующие белки после мануальной классификации субнабора изображений.
Для настоящего анализа количественной пространственной протеомики необходима классификация паттернов для дополнения сегментации органелл. Его разработка обещает существенный прогресс в этом контексте, включая convolutional сети (такие как U-Net136), появление обобщенных моделей делает возможной сегментацию неоднозначных изображений137 и синтез in silico реалистичных флуоресцентных меток из изображений, лишенных меток, таких как прогнозирование меток, определяющих клеточное состояние (мертвые или живыеe) и субклеточные свойства, такие как расположение органелл138,139. Такие успехи компьютерного видения в клеточной биологии указывают на будущие направления, когда клеточные структуры предсказываются на базе лишенныхх меток изображений, что делает возможным многопараментное препарирование клеток.
Способ классификации становится всё более сложным при исследовании сравнительной простарнственной протеомике ув разных типах клеток или при нарушениях, это требует, чтобы изменения в морфологии могли быть различными de facto при изменении расположения белка. Первоначальной целью было определение пространственных различий между выборками скорее, чем между определенными метками, unsupervised компьютерный подход соответствует этой цели. Разработки в этом направлении включают unsupervised clustering модель, способную идентифицироватиь изменения положения белков между 6 разными клеточными компартментами во всех доступных библиотеках дрожжевых GFP 68, а обобщенная модель способна классифицировать паттерны в отличающихся др. от др. с использованием изображений HPA Cell Atlas 15. Фактически эти два исследования, а также большинство др. исследований субклеточных паттернов изображений в классификационных работах, использовали коллекции изображений расположения дрожжевых GFP 19,65,67,74 или коллекции HPA Cell Atlas 52, иллюстрируя, что наборы этих данных являются ценными benchmark ресурсами для разработки улучшенной модели с машинным обучением. Чтобы осуществить мета-анализ локализации белков и успешно разработать классификаторы изображений, необходимы в открытом досупе хранилища изображений 140.
Global comparative spatial proteomics
Субклеточная локализация белков контролируется чётко и многие клеточно-биологические процессы используются для перемещения белков между компартментами. Более того, многочисленные болезни ассоциируют с нарушениями субклеточной локализации белков 4-7. В принципе, глобальная сравнительная пространственная протеомика предоставляет идеальный инструмент для выявления этих физиологических и патологических транслокаций белков на системном уровне. Этот подход беспристрастен и целостный и поэтому подходит для выявления новых и неожиданных аспектов клеточной биологии. Однако, эксперименты с глобальной сравнительной пространственной протеомикой технически сложны.
Challenges and general considerations
Доступные на сегодня методы глобальной пространственной протеомики предоставляют статический 'снимок' клеточных карт скорее, чем непрерывную информацию о динамике белков. Следовательно, все изменения локализации д. быть выведены путем сравнения карт, полученных прежде и после стимулов, генетических альтераций или пертурбаций. Эти ограничения накладывают многочисленные технические и концептуальные ограничения. Успешность сравнительных исследований стала возможной лишь недавно 19-21,24-27,67,73.
Reproducibility
Поскольку обнаружение изменений в субклеточной локализации белков требует сравнения, по крайней мере, двух наборов данных, то воспроизводимость является важной.
Все подходы по MS профилированию нуждаются в частичном разделении органелл с помощью определенной формы градиента центрифугирования. Однако, практически невозможно достичь идентичных фракцией в градиенте во множественных экспериментах и имеются легкие отличия между репликами, и это объясняет, почему комбинирование нескольких наборов данных профилирования улучшает разрешение20,44. Более того, эксперименты с пертурбациями могут влиять на размер или плотность органелл и тем самым сильно изменять паттерны их фракционирования. MS измерения также добавляют экспериментальные шумы. Наконец, благодаря стохастическому элементу разброс (shotgun) MS, наборы белков, идентифицируемые в каждом эксперименте, обычно не идентичны, снижают перекрываемость между биологическими повторами эксперимента и условиями воздействия. Единственным решением этих проблем, помимо технической оптимизации, является проведение множественных повторяющихся сравнительных экспериментов для идентификации действительных изменений21.
Большинство исследований по пространственной протеомике используют библиотеки меченных дрожжей19,67,68,73,97,98,143, тогда как сравнительные исследования изображений протеома человека ограничены одиночными интересующими компартментами144. Для всех базирующихся на изображениях сравнительных исследований, наибольшим затруднением является точность оценки субклеточных локализаций или вручную или с помощью компьютера, и обнаружение отличий между bona fide изменениями положения белков и морфологическими изменениями.
При MS-based и imaging-based подходах др. общая проблема хаключется в том, что две плохо различимые органеллы могут приводить к случайным и поэтому противоречивым оценкам, приводя к ложным положительным результатам.
Types of translocation and their detection
Простейшим типом транслокации является полный переход белка из одного компартмента в др. Однако, доказательства указывают на то, что транслокации часто лишь частичны. Более того, многие белки имеют сложное субклеточное распределение перед и/или после пертурбаций, а изменения в расположении могут согласовываться с количественными скорее, чем качественными сдвигами. Наконец, белок может менять расположение внутри родного и того же компартмента.
Базирующаяся на MS пространственная протеомика дает два типа данных для каждого белка - сырой (численный) профиль количественного распределения и (качественное) предсказание локализации в компартменте, исходя из профиля. Используя изменения в предсказываемой локализации, для идентификации транслоцирующихся белков, это сильно ограничивает возможности анализа, т.к. только полные переходы могут быть обнаружены. Напротив, выявление сдвигов в данных сырого профилирования является более содержательным, особенно для частичных транслокаций и поэтому предпочтительнее, чем предсказуемые локализации21. Интерпретация выявляемых сдвигов д. быть следующей независимой ступенью.
В базирующейся на изображениях пространственно протеомике сначала производится оценка локализации в компартменте, которая также не чувствительна к детекции частичных транслокацией в присутствии мощных (т.е., single majority location) классификаторов. Чтобы осуществить это, несколько исследований использовали мягкие классификаторы (т.е., multiple compartment assignments with likelihood scores) или множественные измерения количественных признаков, это концептуально сходно с профилями, получаемыми с помощью MS-based пространственной протеомики. Т.о., данные изображений становятся ответственными за статистическую детекцию локализации различий, базируясь на различиях профилей 19,68,73.
Determination of false discovery rates
Эксперименты по сравнительной пространственной протеомике, как известно, склонны к продукции высокой пропорции ложно положительных результатов 44; подсчеты false discovery rate (FDR) транслокаций поэтому важны для правильной интерпретации полученных результатов. Наилучший подход - это пробный контрольный эксперимент (control versus control, with no changes expected) в дополнение к действительному эксперименту с пертурбациями и к субъекту, оба при анализе источника информации тех же самых данных 21,24. В этом случает получаются хорошие подсчеты FDR, указывающие, что экспериментальные шумы сходы при обоих условиях. Все опубликованные исследования подтверждают, что повторные эксперименты важны для достижения низких уровней FDR 20,21,24.
Time-course experiments
MS-based динамическое картирование и базирующиеся на антителах изображения являются от природы методами периодического производства (discontinuous methods), т.к. биохимическое фракционирование разрушает клетки, а фиксация убивает клетки. Следовательно, чтобы получить временную информацию о пространственных изменениях, необходима серия экспериментов по сравнительному картированию, скорее всего, записи серии статических изображений для генерации видео 24. Каждая временная точка добавляется поступательно к общим требованиям MS измерений, что обычно является узким горлом для MS-based протеомики. Скрининг библиотеки флуоресцентных белков может, в принципе, предоставлять непрерывную информацию по динамике белка и продуцировать очень впечатляющие резуольтаты в исследованиях, ограниченных одиночным компартментом 22,145. Для геномных библиотек флуоресцентных белков на сегодня исследования всё ещё ограничены статическими снимками (snapshots), хотя и с высоким временным разрешением 73.
Successful applications
Comparative MS profiling
Динамичный подход к картированию органелл является на сегодня наиболее активно используемым методом для сравнительного MS профилирования 21 (rev.146). Он был специфически разработан для сравнительного использования, со специфическим упором на устойчивость производственного процесса и на разработку статистических основ для детекции транслокаций из данных первичного профилирования. Метод впервые был использован для картирования перемещений белков в ответ на стимуляцию epidermal growth factor (EGF) 21 (Fig. 4a). Многочисленные новые и известные транслокации, такие как перемещение EGF рецептора (EGFR) из плазматической мембраны в эндосомы, рекрутирование главных сигнальных адапторов и снование транскрипционных факторов в и из ядра, где были идентифицированы с высокой специфичностью. Уникальным дополнительным свойством этого анализа стало включение количественных оценок белка, предоставление подсчетов ряда копий из наблюдаемых движений. С тех пор метод был использован дважды как инструмент для открытия механистических основ врожденных нарушений. Комплекс адапторного белка 4 (AP-4) необходим для формирования транспорта пузырьков с неизвестной функцией. Генетические дефекты в AP-4 вызывают синдром дефицита AP-4, который характеризуется тяжелой умственной отсталостью и прогрессирующей параплегией. При использовании динамического картрирования органелл к клеткам, дефицитным по AP-4 26, было показано, что AP-4 пузырьки обеспечивают клеточное распределение белка аутофагии ATG9A, который является критическим для биогенеза аутофагосом (и тем самым для поддержания нейронов), в результате открыт новый путь для пространственного контроля аутофагии и, скорее всего, он является причиной синдрома дефицита AP-4 (Fig. 4b). Удивительно, этот подход к картированию идентифицировал ATG9A в качестве непосредственного груза AP-4 пузырьков помимо 4000 профилированных белков, при этом отсутствовала ложно положительная идентификация и без необходимости знания о функции AP-4. Использование этого подхода ьакже использовано при выяснении функции, связанной с AP-5 комплексом 27.
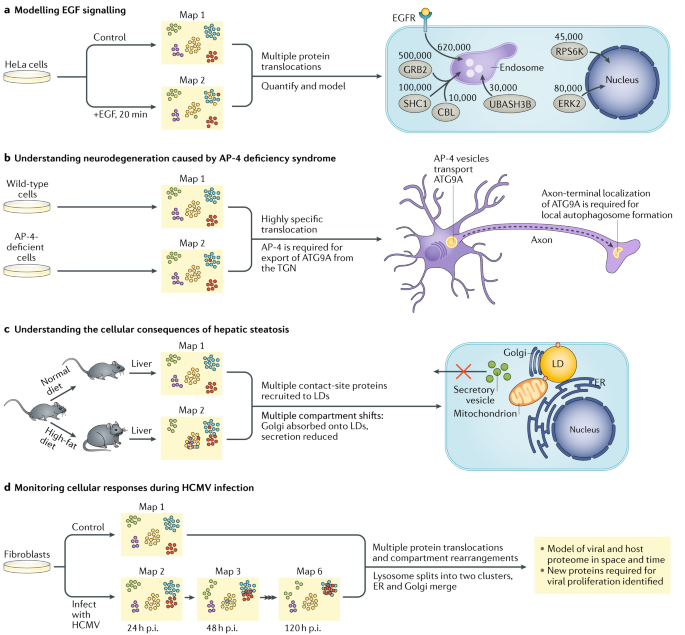 Fig. 4: MS-based comparative spatial proteomics: example applications.
Fig. 4: MS-based comparative spatial proteomics: example applications.
a | Modelling epidermal growth factor (EGF) signalling21. Dynamic organellar maps were generated for HeLa cells that were either untreated or stimulated for 20?min with EGF. Numerous protein translocations were detected, including endocytosis of the EGF receptor (EGFR), recruitment of signalling proteins (GRB2 and SHC1) to EGFR and nuclear shuttling of downstream target transcription factors (ERK2 and RPS6K). Incorporating absolute protein quantification enabled estimation of the copy numbers of proteins undergoing the detected translocations per average cell. Only selected changes are illustrated. b | Understanding neurodegeneration caused by adaptor protein 4 (AP-4) complex deficiency syndrome26. Mutations in the genes encoding subunits of AP-4 cause severe intellectual disability, axonal degeneration and spastic paraplegia by an unknown mechanism. Application of dynamic organellar maps revealed that the key autophagy protein ATG9A is specifically retained at the trans-Golgi network (TGN) in AP-4-deficient cells. An emerging model of the disease is that, in neurons, AP-4 mediates selective transport of ATG9A from the TGN to the distal axon to regulate local autophagosome formation, which is crucial for neuronal homeostasis. c | Understanding the cellular consequences of hepatic steatosis25. Mice were fed a normal diet or a high-fat diet to induce hepatic steatosis, which is a hallmark of non-alcoholic fatty liver disease (NAFLD). Protein correlation profiling maps were generated to monitor the subcellular translocations of proteins in the liver. Key observations include the relocalization of many contact-site proteins to lipid droplets (LDs), the adsorption of the Golgi onto LDs and ensuing secretion defects, which provide a potential molecular mechanism for the reduced hepatocyte function in NAFLD. d | Monitoring cellular responses during human cytomegalovirus (HCMV) infection24. Human primary fibroblasts were infected with HCMV. Organellar rearrangements were followed using localization of organelle proteins by isotope tagging (LOPIT) maps every 24 h over a time course of 120?h. This analysis revealed individual protein translocations that are relevant to viral proliferation and organellar rearrangements, such as the merging of the endoplasmic reticulum (ER) and Golgi and a split of lysosomes into two populations. p.i., post-infection; UBASH3B, ubiquitin-associated and SH3 domain-containing protein B. Part a adapted with permission from ref.21, CC-BY-4.0.
Сравнительное PCP было использовано для детекции изменений в органеллах клеток печени, вызванных диетой с высоким содержание жира25, чтобы охарактеризовать паталогические изменения, происходящие во время печеночного steatosis, являющегося характерным признаком non-alcoholic fatty liver disease (NAFLD) (Fig. 4c). Субклеточное расположение ~4500 белков было картировано и несколько сотен событий транслокаций, предоставив доказательства удивительных перестроек органелл, вызываемых жирной диетой. Напр., аппарат Гольджи оказывался адсорбированным в липидные капельки, а секреция была существенно снижена. Многие белки контактных сайтов органелл также перенаправлялись в липидные капельки, вызывая в результате усиление контактов между липидными капельками и митохондриями. Более того, авт. накладывали профили субклеточного распределения фосфорилированных белков на соотв. профили белков. Т.к. PCP способно оценить каждый белок с множественной субклеточной локализацией, то были установлены события специфичного для органелл фосфорилирования на индивидуальные белки.
Сравнительный LOPIT был использован для отслеживания клеточных перестроек, которые возникают во время инфекции HCMV24. С течением времени 5 LOPIT карт в течение 120 ч отслеживали субклеточную локализацию свыше 4000 хозяйских и 100 вирусных белков (Fig. 4d). Этот анализ выявил основное моделирование органелл, такое как расщепление лизосомами на два отдельных пула и слияние ER и Golgi. Кроме того, авт. идентифицировали многочисленных кандидатов на роль транслокационных событий, это привело к открытию неконвенционного миозина MYO18A в качестве нового фактора, необходимого для эффективной репликации HCMV.
Конечно, в исследованиях, описанных выше 24-27, глобальное картирование MS органелл использовало инструмент чувствительного и специфичного скрининга для установления кандидатов для ортогональной оценки с помощью экспериментов по целенаправленному картированию. provide candidates for orthogonal validation by targeted imaging получению изображений. Этот характерный для определенного класса процесс проявляется как новая стратегия для систематического анализа биологических процессов в клетке.
Comparative imaging-based spatial proteomics
Нацеленные на гены подходы идеально подходят для сравнительной протеомики благодаря простоте анализа множественных пертурбаций, как только будут созданы библиотеки меченных флуоресцентных белков. Принимая во внимание, что такие библиотеки базируются на библиотеках меченных белков, большинство сравнительных исследований использовало S. cerevisiae коллекцию слитых GFP 65 или др. дрожжевые коллекции 98. Некоторые исследования изучали изменения расположения в ответ на условия средовых стрессов, таких как стресс ДНК репликации, индуцированный с помощью methyl methanesulfonate 73,74,143 и hydroxyurea 19,73,74, или в ответ на dithiothreitol (DTT), hydrogen peroxide, nitrogen starvation 67 и rapamycin 19. Напр., 254 белков были найдены как меняющие позицию после повреждения ДНК 73,74, включая неожиданное открытие 20 метаболических энзимов, проникающих в ядро; 235 белков меняли позицию после воздействия или DTT или hydrogen peroxide или после азотного голодания 67. Некоторые из этих исследований базируются исключительно на ручной оценке локализации белков 67,74, тогда как др. использовали компьютерные подходы 19,68,73. Эти исследования независимо пришли к заключению, что изменение позиции белков является широко используемой клеточной стратегией, чтобы бороться со средовыми стрессами и что пропорция протеома, которая меняет положение, сходна с пропорцией, обнаруживающей изменения в численности (Fig. 5a; rev.75). Кроме того, эти два типа реакций в основном не перекрываются в отношении затрагиваемых белков
19,67,74.
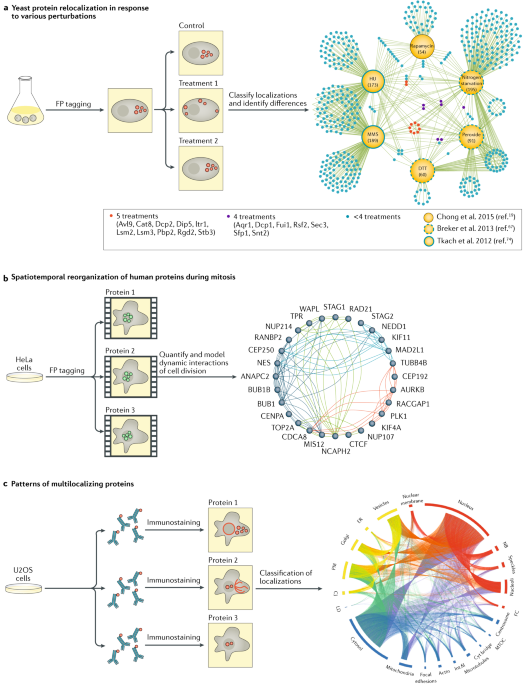 Fig. 5: Imaging-based comparative spatial proteomics: example applications.
Fig. 5: Imaging-based comparative spatial proteomics: example applications.
a | Yeast protein relocalization during stress19,74,100. Proteome-wide imaging-based studies of GFP-tagged proteins in budding yeast revealed patterns of protein relocalization in response to six environmental stress conditions. The network graph summarizes work from three studies; each edge connects a protein relocalization (cyan, purple and red nodes) to the corresponding perturbation (yellow node). Node colour represents the number of conditions in which the relocalization occurs (proteins indicated in parentheses relocalize in response to all treatments). b | Spatiotemporal organization of human proteins during mitosis144. 4D imaging integrated with quantitative protein concentration measurements of 28 GFP-tagged human proteins generated an integrated model of spatiotemporal protein reorganization during mitosis in HeLa cells and enabled analysis of dynamic interactions between the proteins. The network graph shows protein colocalizations during cytokinesis (a subphase of mitosis). Each node is a protein, and each edge corresponds to a dynamic localization cluster. The edge colour corresponds to a localization cluster assigned by computational image analysis. c | Patterns of multilocalizing proteins in various human cell types52. Proteome-wide immunofluorescence analysis of human protein localization across many different cell lines revealed that >50% of proteins localize to multiple cellular compartments. The network graph shows all detected proteins sorted by compartment and organized by meta-compartment (nucleus (red), cytoplasm (blue) and secretory pathway (yellow)). Proteins detected in multiple compartments are connected with an edge, revealing distinct patterns of multilocalization, including between compartments in close proximity (such as nucleoli and nucleoplasm), between compartments that are separated by a membrane (such as the nucleoplasm and cytosol) and between more distant compartments (such as proteins on the plasma membrane and those in the nucleoplasm). CJ, cell junction; Cyt bridge, cytokinetic bridge; DTT, dithiothreitol; ER, endoplasmic reticulum; FC, nucleoli fibrillar centre; FP, fluorescent protein; HU, hydroxyurea; LD, lipid droplet; MMS, methyl methanesulfate; NB, nuclear body; PM, plasma membrane. Part areproduced with permission from ref.75 (Torres, N. P., Ho, B. & Brown, G. W. High-throughput fluorescence microscopic analysis of protein abundance and localization in budding yeast. Crit. Rev. Biochem. Mol. Biol. 51, 110-119 (2016)), Taylor & Francis Ltd (http://www.tandfonline.com). Part b adapted from ref.144, Springer Nature Limited. Part cadapted with permission from ref.52, AAAS.
Чтобы получить больше информации о перемещениях белков проводился компьютерный мета-анализ 24 дрожжевых скринов104 (представленных 400000 изображений). По этой причине был использован метод детекции unsupervised, компьютерной локализации изменений147, после визуальной интерпретации воздействий (hits). Этот подход позволил авторам различать реакции, нацеленные на специфические пертурбации, от более генерализованных реакций и идентифицировать группы белков со сходными паттернами релокализации, указывающими на функциональные связи. Более того, авт. продемонстрировали, что хокогда расположение некоторых белков меняется это сопровождается изменениями транскрипции или количества белков, субклеточной локализации, что в общем не зависит от сложности регуляции.
Плодотворное исследование митозов в клетках человека с использованием данных по 4D изображений для анализа пространственного распределения белков со временем, совместно с 3D концентрационными данными для 28 белков человека, которые были genomically (homozygously) нагружены улучшенным GFP144. Каноническая модель пространственно-временной реорганизации белков во время митозов в клетках человека была создана с использованием компьютерного анализа изображений и данных по интеграции (Fig. 5b). Это исследование продемонстрировало мощный generic подход к stochiometric анализу простраственно-вренменного перераспределения белков и проложило путь к сходным исследованиям д. клеточных процессов.
HPA Cell Atlas картировал расположение 12003 белков человека в нормальных состояниях в 30 разных клеточных структурах 52. Эта работа показала, что ~50% (6163) белков расположены в двух или более клеточныхъ компартментах. Путем картирования паттернов множественных локализаций внутри клетки (Fig. 5c), это исследование продемонстрировало, что некоторые органеллы, такие как митохондрии и ER, в основном содержат белки, которые расположены исключительно в этих органеллах, тогда как др., такие как плазматическая мембрана и ядро, содержат мнжество белков с локализацией во многих местах; эти находки были оценены с использованием hyper-LOPIT. Локализация каждого из белков была проанализировна в трех разных линиях клеток человека, это позволило установить, что локализующиеся во многих местах белки, скорее всего, обнаруживают пространственную изменчивость между клеточными линиями. В целом, 3546 multilocalizing белков обнаруживают зависимую от линии клеток локализацию.
Future directions
Recent advances in genomics, transcriptomics, proteomics and other omics technologies have provided unprecedented systems biology tools and drastically reshaped our understanding of cell biology. However, the spatial organization of the cell is not directly assayed by any of these approaches, although evidence suggests that dynamic subcellular reorganization represents an independent layer of regulation as important as changes in abundance at the RNA and protein levels. The systematic application of spatial proteomics will thus be essential for a complete understanding of cellular physiology. Until recently, large-scale proteomics has been the domain of a few specialist laboratories, but this is changing rapidly. The required technology (that is MS or imaging) is becoming more widely available, as are user-friendly software solutions for data analysis. Consequently, spatial proteomics experiments have become fairly accessible, and we encourage readers outside the field to join in (Supplementary Box 1; Supplementary Box 2).
Multiple factors make the cellular proteome much more complex than expected from a gene count (Box 2). Spatial proteomics is ideally poised to unravel the functional importance of this intriguing cellular complexity, and we expect that it will have a major role in doing so. For example, mapping proteins that localize to multiple compartments will be a key step towards understanding organelle crosstalk, interlinked cellular processes and identifying proteins with moonlighting activity. Mapping proteins with temporal and spatial variability in single cells will add important insights into cellular signalling dynamics. Finally, elucidating the biological roles of proteoforms (that is, splice variants and post-transcriptionally modified forms of proteins) is a central yet largely uncharted aspect of cellular complexity and organization, which can be explored using spatial proteomics.
As we outline here, imaging-based and MS-based approaches offer complementary insights into the spatial proteome (Supplementary Table 1; Supplementary Box 2). Importantly, both approaches are required to fully understand cellular complexity, and several studies have demonstrated the strong synergy obtained by combining MS-based spatial proteomics with imaging52,55,124.
A further important (and we think imminent) advance will be the combination of spatial proteomics with other omics technologies. As an example, transcriptomics can provide cell-specific expression databases, including cell-type-specific splice variants or single nucleotide polymorphisms, thus empowering proteomics to discover novel proteoforms (proteogenomics148). In combination with metabolomics, organellar rearrangements can be functionally linked to changes in metabolism25. Similarly, in conjunction with RNA sequencing, spatial proteomics has the potential to connect mRNA biology with subcellular localization149,150,151. In our opinion, spatial proteomics should become an orthogonal and integrated technology in cell mapping efforts such as the Human Cell Atlas, which aims to characterize all human cell types152.
Finally, a major future challenge will be the collection and integration of the various data sets produced in past and future spatial proteomic studies. At present, there is little crosstalk between major resources such as the HPA Cell Atlas52 and individual studies that also provide subcellular localization databases20,21,24,25,47,50. Given the dynamic nature of protein spatial organization, repositories for both imaging-based and MS-based spatial proteomics data need to be developed to enable meta-analysis of studies using different cell types, perturbations and growth conditions. Existing repositories, such as UniProt, should consider how to incorporate and cross-reference these data sets for the benefit of all cell biologists.
In conclusion, we envisage a new era of cellular modelling in which the spatial dynamics of proteins are integrated with other omics measurements to gain insight into the crosstalk between the different layers of cellular regulation, leading to a greater understanding of cellular phenotype and activity.
Box 2 Complexity of the cellular proteome: future challenges and opportunities for spatial proteomics вжж
Multiple factors make the cellular proteome much more complex than predicted from a mere gene count. Spatial proteomics is ideally poised to unravel the functional importance of this intriguing cellular complexity.
Multilocalizing and 'moonlighting' proteins
Several studies indicate that ~50% of the human proteome is localized to multiple cellular compartments52,65. This observation raises many questions, such as whether the different protein populations are static or dynamic, how their cellular sorting is achieved, whether the different populations correspond to different proteoforms and, most importantly, whether these proteins have context-specific functions (that is, moonlighting activity). Moonlighting is defined as proteins having two or more different cellular functions that do not result from genetic variations, RNA splicing or pleiotropic effects153,154 but result from differences in subcellular localization, substrates, oligomerization or post-translational modifications (PTMs)155. The number of known moonlighting proteins is rapidly increasing. Databases list nearly 700 proteins that have two characterized independent functions156,157,158, and it has been estimated that 23% of the human proteome is moonlighting159. A remarkable 78% of the known moonlighting proteins are involved in disease, and 48% are current drug targets156, emphasizing the importance of moonlighting for cellular function. Imaging-based spatial proteomics will be a key technology for the identification of multilocalizing proteins to provide a better understanding of their crucial role in spatial coordination of the complex adaptive systems that underlie cellular functionality.
Proteoforms
Each molecular form of a protein is called a proteoform 160, including variability from the DNA sequence (135000 coding single nucleotide polymorphisms (SNPs) validated in UniProt 161), RNA splicing (93% of human genes undergo RNA splicing 162) and diverse PTMs, such as phosphorylation, ubiquitylation, alkylation and glycosylation. A typical human cell is estimated to contain 6 million coexisting proteoforms 163; although this number is far below the theoretically possible number of combinations 164(reviewed elsewhere 165), it reveals the massive complexity of the proteome. Studies aimed at exploring the functional consequences of alternative splicing have shown that it frequently leads to inclusion or exclusion of targeting sequences, such as membrane anchors or mitochondrial signals, and that the sets of interaction partners of different isoforms of a given protein are often as different as those of proteins encoded by different genes 166. Furthermore, many PTMs, especially phosphorylation and ubiquitylation, are known to regulate protein subcellular localization. Conceptually, mass spectrometry (MS)-based spatial proteomics, aided by proteogenomics 148, can provide insights about proteoform nature and subcellular localization and will provide a key technology to understand the largely unexplored functional significance of proteoform complexity.
Protein abundance
Cellular protein abundance spans seven orders of magnitude in human cells18. To model cellular processes, knowledge of absolute protein copy number per cell and its changes is required. Both MS-based and imaging-based spatial proteomics can provide this information.
Single-cell variability
It is becoming increasingly clear that populations of genetically identical cells show variability in protein expression52,66, for example, during cell differentiation 69,70 and in response to environmental fluctuations 67,69 or drugs 72. Our understanding of the consequences of this cellular heterogeneity is still rudimentary. Imaging-based methods will likely have a key role in the elucidation of the causative factors, such as stochasticity, microenvironments, cellular states such as the cell cycle or population bet-hedging strategies. Of note, workflows for single-cell proteomic analysis are currently emerging 167.
Protein dynamics
Protein subcellular localization is tightly controlled, and many proteins change localization in response to a stimulus, a perturbation or in disease. Global comparative spatial proteomics, either imaging-based, interaction-based or MS-based, provides an ideal tool to capture these physiological and pathological protein translocations at the systems level and should become a widespread discovery tool for cell biologists.
|