Когда животное сталкивается с угрозой в окружающей среде или другими стрессовыми стимулами, оно должно быстро мобилизовать запасы энергии для поддержания соответствующего защитного поведения, такого как побег, бросок или замирание1,2. Быстрая мобилизация энергии стимулирует сердечно-сосудистую и мышечную реакцию на стресс и способствует более активному распределению когнитивных ресурсов, необходимых для обработки контекстуальной информации, связанной с угрозой3-5. Параллельно необходимо подавлять поведенческие стереотипы, которые могли бы конкурировать с защитными стратегиями, такими как поиск пищи и поглощение7. Реакция гликемии на стресс коррелирует с поведением "сражайся и убегай"8, а инфузии глюкозы вызывают увеличение сердечного выброса и кровяного давления, подобное стрессу, 9,10, в дополнение к облегчению кодирования пространственных воспоминаний11-13. Эти метаболические адаптации, вызванные угрозой, в значительной степени сохраняются у разных видов, что позволяет предположить, что они обеспечивают значительное эволюционное преимущество14. Несмотря на их биологическую важность, на удивление мало известно о мозговых цепях, которые управляют адаптивными реакциями на гипергликемию и гипофагию при остром стрессе15.
Здесь мы выявили новую миндалевидно–гипоталамо–печеночную ось, которая регулирует быструю метаболическую адаптацию к острому стрессу с помощью механизмов, не зависящих от классической адреномедуллярной и гипоталамо–гипофизарно–надпочечниковой (HPA) стрессовых систем. Мы демонстрируем, что повторяющийся стресс нарушает работу миндалевидного тела и печени, вызывая связанные со стрессом метаболические нарушения, тем самым связывая хронический стресс с метаболическими нарушениями, такими как сахарный диабет 2 типа (T2D).
Acute stress rapidly changes metabolism
Чтобы исследовать основные механизмы метаболической адаптации, связанной со стрессом, мы сначала выявили острые стрессоры, которые повышали уровень глюкозы в крови и подавляли аппетит у мышей C57Bl/6. Острый ограничительный стресс быстро повышал уровень глюкозы в крови и нарушал толерантность к глюкозе у мышей, которые были ограничены в пище в течение 6 часов перед тестированием (рис. 1а, б, расширенные данные рис. 1а, в и дополнительный рис. 1н). Ограничительный стресс также повышал уровень кортикостерона в плазме крови (рис. 1с), что согласуется с активацией оси HPA. Уровни адреналина, глюкагона и глицерина в плазме крови также повышались в результате ограничительного стресса (расширенные данные на рис. 1д, д, г), что согласуется с активацией адреномедуллярной стрессовой системы, стимулирующей симпатическую активность надпочечников, поджелудочной железы и жировой ткани. Напротив, уровни инсулина и норадреналина в плазме крови у этих мышей не изменились под воздействием ограничительного стресса (расширенные данные, рис. 1c, f). Известно, что ограничение питания вызывает метаболическую адаптацию, подобную стрессу, включая снижение уровня инсулина в плазме крови и повышение выработки глюкозы печенью 16. Поэтому мы также оценили метаболические реакции на стресс у мышей, получавших полноценное питание. Острый стресс у мышей, получавших полноценное питание, повышал уровень глюкозы в крови, глюкагона и кортикостерона в плазме крови и снижал уровень инсулина в плазме крови (дополнительный рисунок. 1a–d). Ограничительный стресс также увеличивал экспрессию глюкозо-6-фосфатазы (G6pc) в печени без влияния на фосфоенолпируваткарбоксикиназу (Pck1) или содержание гликогена в печени у мышей, которых кормили (рис. 1d и расширенные данные рис. 1h), что согласуется со стресс-индуцированным увеличением способности печени вырабатывать глюкозу. Социальный стресс, вызванный воздействием на мышей запаха самца-конспецифика в изолированной клетке, аналогичным образом повышал уровень глюкозы в крови и нарушал толерантность к глюкозе (Расширенные данные, рис. 1i–k и дополнительный рис. 1l). Стресс, вызванный локализацией клеток, повышал уровень кортикостерона в плазме крови и приводил к незначительному увеличению экспрессии G6pc и Pck1 в печени, а также к неизменному уровню адреналина в плазме, гормонов поджелудочной железы и глицерина (рис. 1). 1l–r,t). Стресс, связанный с территорией в клетке, также подавлял потребление пищи (расширенные данные, рис. 1с). Даже кратковременного (5 минут) ограничения или стресса, связанного с территорией в клетке, было достаточно, чтобы повысить уровень глюкозы в крови и кортикостерона в плазме (дополнительный рисунок). 1e–g, i–k), что согласуется с быстрой мобилизацией глюкорегуляторных механизмов, которые обеспечивают метаболическую адаптацию к физическим и социальным стрессорам.
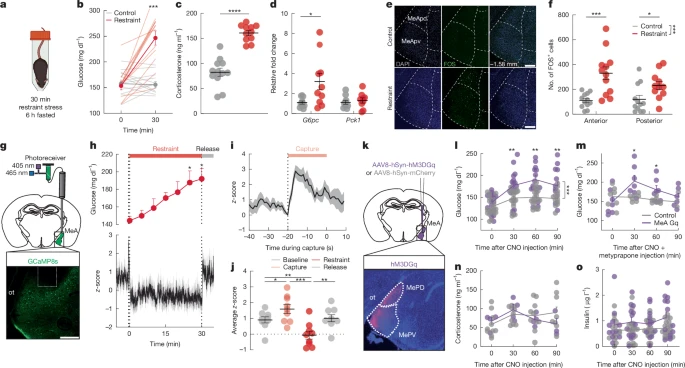 Рис. 1. Стрессовая активация нейронов MeA для регуляции уровня глюкозы.
Рис. 1. Стрессовая активация нейронов MeA для регуляции уровня глюкозы.
Stress activates MeA neurons
Известно, что социальные стимулы изменяют активность нейронов MeA17, а внешние и внутренние сенсорные сигналы, связанные с угрозой, сходятся в MeA18-20. Нейроны MeA посылают сигналы в области мозга, чувствительные к стрессу, которые регулируют эндокринные, вегетативные и метаболические процессы, включая ядра гипоталамуса21 и ядро ложа терминального полосатого тела22 (BNST). Таким образом, мы предположили, что нейроны MeA участвуют в метаболической адаптации к острому стрессу. Сдерживающий стресс увеличивал количество иммунореактивных клеток FOS (FOS+) в передней и задней областях, а также в дорсальной и вентральной областях MeA по сравнению с контрольными мышами, не подвергавшимися стрессу, и по сравнению со стресс-индуцированными клетками FOS+ в соседней базолатеральной миндалине (BLA) и центральной миндалине (CEA) ядра (рис. 1e,f и расширенные данные на рис. 2a–c). Используя кальциевую визуализацию in vivo, основанную на фотометрии, мы обнаружили, что нейронная активность в MeA быстро повышалась в течение "периода захвата", 20-секундного периода перед удержанием, когда исследователь вручную удерживал мышь, а затем помещал ее в удерживающую трубку на 5 или 30 минут удерживающего стресса (Рис. 1g–j и дополнительный рис. 2a–c). Активность MeA была подавлена в течение периодов иммобилизации у мышей, но возвращалась к исходному уровню, когда мышей освобождали от иммобилизации (рис. 1g–j и дополнительная рис. 2a–c). Воздействие на мышей специфического запаха в изолированной клетке в течение 5 или 30 минут вызывало быстрое повышение активности MeA с постепенным возвращением к исходному уровню (рис. с расширенными данными 2d–f и дополнительный рис. 2d–f). Примечательно, что вызванное стрессом ограничение свободы и территориализация клеток повышение активности MeA происходило до повышения уровня глюкозы в крови (рис. 1h, Расширенный рисунок данных 2d и дополнительный рисунок 2a, d). Аналогичным образом, активность MeA была значительно повышена при воздействии физического стресса, толчка ногой и визуального стресса, приближении ‘robobug’23, оба из которых значительно повышали уровень глюкозы в крови (дополнительный рисунок. 2g–l). Напротив, активность MeA существенно не изменялась в новой чистой клетке и не была связана с изменениями двигательной активности в условиях отсутствия стресса, что позволяет предположить, что нейроны MeA не кодируют движение как таковое (дополнительный рисунок. 2m–r). Эти данные свидетельствуют о том, что нейронная активность в MeA активизируется, когда мыши пытаются уклониться от угрожающих стимулов, и совпадает с быстрой мобилизацией энергетических резервов для поддержания такого адаптивного поведения.
MeA neurons regulate glucose and feeding
Затем мы использовали хемогенетику для изучения участия MeA в метаболической адаптации к стрессу. Мы ввели адено-ассоциированный вирус, экспрессирующий возбуждающий hM3DGq (AAV-hSyn-hM3DGq-mCherry), DREADD (дизайнерские рецепторы, активируемые исключительно дизайнерскими лекарственными средствами), или контрольный вирус (AAV-hSyn-mCherry) в MeA мышей (рис. 1k). Инъекция Clozapine-N-oxide (CNO, 3 мг/кг-1, внутрибрюшинная инъекция) значительно повышала уровень глюкозы в крови у контрольных мышей, экспрессирующих DREADD без стресса, но не экспрессирующих mCherry (рис. 1l). Анализ гормональных реакций поджелудочной железы, зависящих от пола, выявил значительное повышение уровня инсулина в плазме крови у мышей-самцов, экспрессирующих DREADD, через 90 минут, но не у контрольных мышей, и подавление уровня глюкагона в плазме крови у мышей-самок, экспрессирующих DREADD, по сравнению с исходным уровнем (рис. 1о и расширенные данные на фиг. 2l–n). Учитывая, что активность MeA повышалась под воздействием физических, социальных и визуальных стрессоров, мы были удивлены, обнаружив, что стимуляция нейронов MeA, опосредованная DREADD, повышала уровень глюкозы в крови без существенных различий в содержании адреналина в плазме (расширенные данные на рис. 1). 2h) или кортикостерона в плазме крови (рис. 1n) между hM3DGq и контрольными группами. Действительно, химиогенетическая стимуляция нейронов MeA значительно повышала уровень глюкозы в крови даже в присутствии ингибитора синтеза кортикостерона metyrapone (50 мг/кг-1, подкожная инъекция) (рис. 1м) в дозе, достаточной для того, чтобы притупить вызванное стрессом повышение уровня глюкозы в крови и блокировать вызванное стрессом повышение уровня кортикостерона в плазме (Дополнительный рис. 3k,l). Это говорит о том, что нейроны MeA провоцировали повышение уровня глюкозы в крови без привлечения связанных со стрессом глюко-регуляторных гормонов из коры надпочечников или поджелудочной железы, контролируемых HPA и адреномедуллярной стрессовой системами. Активация MeA–нейронов DREADD у мышей, подвергшихся ограничению или стрессу в условиях территории клетки, или при введении глюкозы, не приводила к дальнейшему усилению реакции на глюкозу (дополнительный рис. 3c-j). В дополнение к повышению уровня глюкозы в крови, стимуляция MeA, опосредованная DREADD, вызывала кратковременное, похожее на стресс, подавление приема пищи в течение первого часа (стандартная лабораторная еда) у мышей, не получавших пищу в течение ночи (расширенные данные, рис. 2к). Стимуляция MeA аналогичным образом подавляла потребление вкусной пищи, обусловленное вознаграждением, у полностью накормленных мышей в течение 2 часов (дополнительный рис. 3b). Напротив, активация MeA не оказывала влияния на защитное поведение, связанное со страхом или тревогой, в приподнятом лабиринте, светлой-темной коробке или аппарате с открытым полем (расширенные данные, рис. 2j). Это говорит о том, что активация MeA приводит к вызванным стрессом изменениям в метаболизме и энергетическом гомеостазе независимо от реакций тревоги и страха.
Чтобы исследовать взаимосвязь между нейронной активностью MeA и метаболическими реакциями с большим временным разрешением, мы исследовали влияние оптогенетической стимуляции MeA на уровень глюкозы в крови. Мы вводили адено-ассоциированный вирус (AAV), экспрессирующий возбуждающий опсин ChR2 (AAV-hSyn-hChR2(H134R)) или контрольный вирус (AAV-hSyn-eGFP), в MeA и имплантировали оптическое волокно непосредственно над местом инъекции. Оптогенетическая стимуляция MeA (470 нм, 20 Гц) в течение 5 мин и 15 мин значительно повышала уровень глюкозы в крови в конце периода стимуляции по сравнению с исходным уровнем у мышей, экспрессирующих ChR2 без стресса, но не у контрольных мышей (фиг. Расширенные данные). 2o–q и дополнительный рис. 3n,o). Оптогенетическая активация не приводила к значительному повышению уровня глюкозы в крови у мышей, подвергшихся стрессу, или в сочетании с контролем уровня глюкозы (дополнительный рис. 3p–y). В соответствии со стимуляцией DREADD, оптогенетическая стимуляция MeA (15 мин) также вызывала резкое подавление потребления пищи у мышей, лишенных пищи, во время стимуляции (дополнительный рис. 3z). В совокупности эти данные свидетельствуют о том, что нейроны MeA активируются при воздействии острых стрессовых факторов, что приводит к метаболической адаптации к стрессу.
MeA circuit responses to stress
Далее мы исследовали механизмы на уровне цепей, с помощью которых нейроны MeA повышают уровень глюкозы в крови и подавляют аппетит.В BNST обнаружены окончания аксонов, экспрессирующие mCherry, что указывает на связь с нейронами MeA. Введение синаптофизина, меченного mCherry (синаптически локализованного белка), в MeA выявило плотные окончания аксонов, экспрессирующих mCherry, в областях гипоталамуса, которые, как известно, регулируют метаболизм, включая медиальную преоптическую область, латеральный гипоталамус и вентромедиальный гипоталамус (VMH) (рис. 2а, б). Мы также обнаружили экспрессирующие mCherry терминалы в BNST, компоненте расширенной миндалины, который, как известно, регулирует физиологическую и поведенческую адаптацию к стрессу
24 (рис. 2а, б). Поскольку цепи, включающие VMH и BNST, участвуют в регуляции уровня глюкозы
25,26 и задействованы стрессовыми стимуляциями
24,27, мы исследовали, регулируют ли нейроны MeA, которые проецируются на VMH (нейроны MeA
VMH) и/или BNST (нейроны MeA
BNST) метаболическую адаптацию к стрессу. Во-первых, мы определили, проецируются ли одни и те же популяции нейронов MeA как на VMH, так и на BNST. Мы ввели ретроградно перемещающийся AAV, экспрессирующий красный флуоресцентный белок (AAVretro-RFP), в BNST и AAVretro, экспрессирующий зеленый флуоресцентный белок (AAVretro-GFP), в VMH (рис. 2в). Мы обнаружили, что менее 12% меченых нейронов совместно экспрессировали как RFP, так и GFP (рис. 2d, e). Это говорит о том, что в значительной степени непересекающиеся популяции нейронов MeA проецируются на VMH или BNST. Затем мы использовали иммуномаркировку FOS в сочетании с отслеживанием AAVretro, чтобы определить, активируются ли нейроны MeA
VMH или MeA
BNST при ограничительном стрессе, который повышает уровень глюкозы в крови. Ограничительный стресс увеличил количество нейронов FOS+ MeA
VMH примерно в два раза без увеличения количества нейронов FOS+ MeA
BNST (рис. 2f, g). Это говорит о том, что острый ограничительный стресс преимущественно увеличивает активность нейронов MeA
VMH.
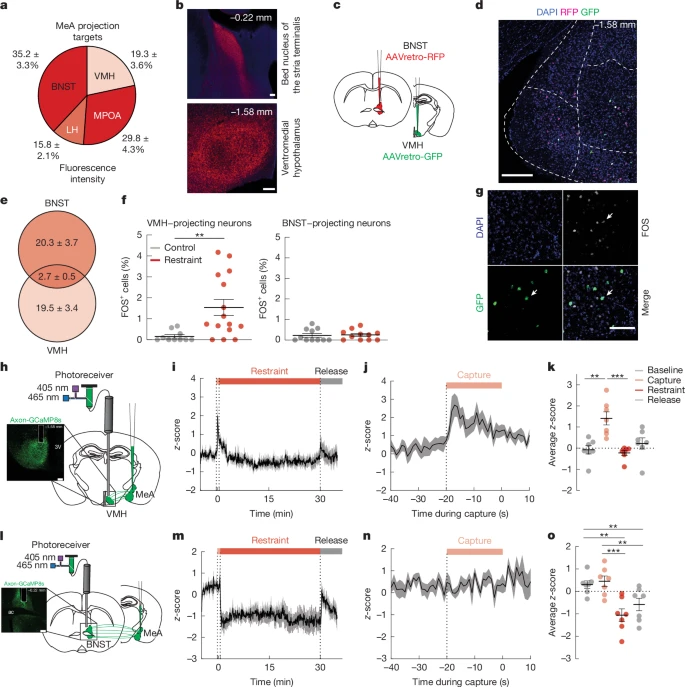 Рис. 2. Острый ограничительный стресс активирует нейроны MeA>VMH, но не нейроны MeA>BNST.
Рис. 2. Острый ограничительный стресс активирует нейроны MeA>VMH, но не нейроны MeA>BNST.
Чтобы расширить эти исследования и определить динамику нейронной активности в этих цепях во времени, мы использовали волоконную фотометрию для визуализации кальция in vivo в проекционных цепях MeAVMH и MeABNST с подачей MeA GCaMP, нацеленного на аксоны (AAV-hSyn-axon-jGCaMP8s-P2A-mRuby3), и размещением волокна над VMH (рис. 1). 2h) или BNST (рис. 2l). Мы обнаружили, что нейронная активность проекционных нейронов MeAVMH была значительно повышена в период capture непосредственно перед 5–минутным и 30-минутным удерживающим стрессом (рис. 2i-k и дополнительный рис. 4a–e). Активность в проекционных нейронах MeAVMH также повышалась при 5–минутном и 30-минутном territorialized cage stress (расширенные данные на рис. 3a-d и дополнительный рис. 4f–h), с использованием footshock и подхода robobug (дополнительный рис. 4m–p), но не с использованием новой чистой клетки (дополнительный рис. 4i–l). Вызванное стрессом повышение активности проекционных нейронов MeAVMH предшествовало повышению системного уровня глюкозы у мышей с непрерывным мониторингом уровня глюкозы (расширенные данные на рис. 3e,f). Напротив, в то время как активность проекционных нейронов MeABNST значительно повышалась при 5–минутном и 30–минутном воздействии новой клетки (расширенные данные, рис. 3g–j и дополнительный рис. 4v-x), активность значительно не повышалась до, во время или после 5-минутного или 30-минутного ограничительного воздействия (рис. 2m-o). и дополнительный рис. 4q–u), с помощью footshock или с использованием метода robobug (дополнительный рис. 4ac–af), или с использованием новой чистой клетки (дополнительный рис. 4y–ab).
Diverse MeA neurons innervate the VMH
Мы стремились идентифицировать нейроны MeA, которые проецируются на VMH и регулируют гипергликемию, вызванную стрессом. Мы применили пространственную транскриптомику (Xenium, 10X Genomics), чтобы охарактеризовать экспрессию библиотеки из 359 генов с маркерами ненейронного и нейронально-клеточного типов, включая известные гены, экспрессируемые MeA[28] (Таблица дополнительных данных 2, S1), для определения генетических фенотипов нейронов MeA
VMH с одноклеточным разрешением. Коронарные срезы, содержащие MeA (от -0,7 мм до -2,06 мм от bregma), были получены от мышей C57Bl/6 с помощью VMH-инъекций AAVretro-hSyn-mCherry для флуоресцентного мечения нейронов MeA, проецирующихся на VMH. Мы проанализировали в общей сложности 21 607 клеток, сопоставленных с MeA, из шести биологических копий (таблица дополнительных данных 2, S2 и S3). Неконтролируемая кластеризация выявила 15 отдельных групп клеток на основе их транскрипционных профилей, выявив основные популяции ненейронных и нейрональных клеток в пределах MeA (рис. 3а и расширенные данные рис. 4а). Транскрипты генов, которые считаются маркерами различных типов клеток, демонстрируют пространственно изменчивую экспрессию в соответствии с клеточной анатомией MeA (рис. 3b). Затем мы более подробно рассмотрели нейронные популяции, сопоставимые с MeA (12 512 нейронов). Нейроны MeA были повторно сгруппированы, в результате чего был получен набор из 20 нейронных кластеров с четким пространственным распределением (рис. 3c, Расширенные данные рис. 4h и таблица дополнительных данных 2, S4). Единственная основная популяция
Vgat (также известная как
Slc32a1), экспрессирующая GABA-ергические (экспрессирующие γ-аминомасляную кислоту) нейроны, была выделена в пространстве с равномерной множественной аппроксимацией и проекцией (UMAP), тогда как три популяции глутамат-ергических нейронов были выделены на основе исключительной экспрессии
Vglut1 (также известного как
Slc17a7) или
Vglut2 (также известный как
Slc17a6) или коэкспрессия обоих генов (
Vglut1/2+) (рис. 3d и расширенные данные рис. 4b–f). Эти нейронные популяции были топографически организованы в MeA, причем GABA-ергические нейроны были распределены в основном по длине дорсальных MeA, а популяции возбуждающих нейронов были сосредоточены в вентральной части MeA, при этом экспрессия
Vglut2 была в основном в передней части MeA, а
Vglut1 – в задней MeA (рис. 3e и расширенные данные рис. 4g-j).. Индуцированная стрессом экспрессия FOS была повышена как по дорсально–вентральной, так и по переднезадней оси MeA (рис. 1f и расширенные данные рис. 2b), что позволяет предположить, что стресс активирует как тормозные, так и возбуждающие популяции нейронов MeA.
 Рис. 3. Экспрессия генов в VMH-проекционных нейронах MeA.
Рис. 3. Экспрессия генов в VMH-проекционных нейронах MeA.
Затем мы зарегистрировали изображения Xenium с помощью флуоресцентного микроскопа для нейронов mCherry+, помеченных AAVretro, идентифицировав проекционные нейроны MeAVMH и ядра (помеченные DAPI). Это позволило выделить 305 нейронов mCherry+, распределенных по длине MeA, причем глутаматергические нейроны составляли 74% этих клеток (рис. 3f, g и расширенные данные рис. 4k, l). Наибольшая доля нейронов mCherry+ MeA присутствовала в кластерах 3 и 4 (Vglut2+), 7 (Vglut1+), 18 (Vglut1/2+) и 11 (GABA-ергических) (расширенные данные на рис. 1). 4l и Дополнительная таблица данных 2, S5). Примечательно, что нейроны mCherry+ MeA были обогащены несколькими генными транскриптами по сравнению с нейронами mCherry- (Расширенные данные на рис. 4m и дополнительные данные в таблице 2, S6 и S7). Аллельные вариации во многих из этих генов, обогащенных MeAVMH, связаны с метаболическими процессами, включая уровень глюкозы в крови, T2D и массу тела, как показано в их таблице генетических данных человека 29 (https://hugeamp.org/) (рис. 3k). Чтобы подтвердить эти транскриптомные данные, мы ввели Cre-зависимый синаптофизин-mCherry в MeA мышей Vglut2-cre и Vgat-cre. Мы обнаружили аксонные окончания, экспрессирующие mCherry, в области VMH как у мышей Vglut2-cre, так и у мышей Vgat-cre (рис. 3h, i), что согласуется как с глутаматергическими, так и с GABA-ергическими нейронами, иннервирующими VMH. Соответственно, хемогенетическая активация глутаматергических и GABA-ергических нейронов MeA (с использованием AAV-CamK2a-hM3DGq-mCherry и AAV-hDlx-GqDREADD-dTomato-Fishell-4, соответственно) значительно повышала уровень глюкозы в крови по сравнению с контрольными мышами (AAV-hSyn-mCherry) (рис. 3j). Эти данные свидетельствуют о том, что MeAVMH-circuit состоит из смешанных популяций глутаматергических и GABA-ергических нейронов, при этом активация как возбуждающих, так и тормозных цепей способствует повышению уровня глюкозы в крови.
MeAVMH neurons regulate blood glucose
Затем мы исследовали, изменяет ли манипулирование активностью контура MeA
VMH метаболические реакции на острый стресс. Хемогенетическое подавление нейронов MeA
VMH было достигнуто путем совместного введения AAVretro-Cre в VMH и AAV-DIO-hSyn-hM4DGi в MeA одних и тех же мышей (рис. 4а). Подавление нейронов MeA
VMH с помощью CNO (3 мг/кг
-1) притупляло гипергликемические реакции, вызванные 30-минутным ограничением и стрессом клетки (рис. 4b и расширенные данные на рис. 1). 5b) и снизили повышение уровня глюкозы при 5-минутном ограничении и территориальном напряжении в клетке (дополнительный рисунок. 5g–o). Напротив, вызванное стрессом повышение уровня кортикостерона, адреналина и глюкагона не изменялось при инактивации нейронов MeA
VMH (расширенные данные на рис. 5d,f, g). Уровни инсулина и норадреналина в плазме крови существенно не отличались между группами (расширенные данные на рис. 1). 5e и дополнительный рис. 5k). Подавление активности нейронов MeA
VMH не приводило к изменению уровня глюкозы в плазме у мышей, не подвергавшихся стрессу, которые голодали в течение 6 часов (расширенные данные, рис. 5с и Дополнительная рис. 5f), или к изменению уровня глюкозы в крови при проведении теста на толерантность к глюкозе (GTT) или теста на толерантность к инсулину (дополнительная фиг. 5p–r). Подавление работы нейронов MeA
VMH также не влияло на потребление пищи мышами натощак или на поведение, похожее на беспокойство, в аппарате открытого поля (дополнительный рисунок. 5s–u). У мышей, экспрессирующих конструкции MeA
VMH, наблюдалось вызванное стрессом повышение уровня глюкозы, кортикостерона и адреналина, сходное с таковым у мышей без инъекции AAV (дополнительный рис. 5c-e).
 Рис. 4: Нейроны MeA, отвечающие за VMH, регулируют уровень глюкозы в крови.
Рис. 4: Нейроны MeA, отвечающие за VMH, регулируют уровень глюкозы в крови.
Затем мы экспрессировали hM3DGq в нейронах MeAVMH (рис. 4в) и оценивали регуляцию уровня глюкозы в крови. У мышей, которых голодали в течение 6 часов, уровень глюкозы в крови повышался за счет хемогенетической стимуляции нейронов MeAVMH (Расширенные данные на рис. 4в). 5i и дополнительный рис. 6b). Уровни кортикостерона, глюкагона, адреналина и норадреналина в плазме крови не изменялись при активации MeAVMH, хотя уровень инсулина в плазме крови повышался через 90 мин, возможно, в качестве компенсаторной реакции на повышение уровня глюкозы в крови (расширенные данные Рис. 5j–m и дополнительные рис. 6j,k). Стимуляция нейронов MeAVMH приводила к стойкому повышению уровня глюкозы в крови в тесте GTT по сравнению с контрольными мышами без существенной разницы в реакции плазмы на инсулин (рис. 4d и дополнительные рис. 6c, h,i). Это говорит о том, что повышенная активность нейронов MeAVMH задерживает восстановление уровня глюкозы в крови до гомеостатического уровня. Более того, активация MeAVMH повышала уровень глюкозы в крови во время GTT в присутствии ингибитора синтеза кортикостерона metyrapone (рис. 4e и дополнительный рис. 6d). Гипергликемия, вызванная ограничениями или социальным стрессом (5 и 30 мин), не была дополнительно усилена стимуляцией MeAVMH нейронов, опосредованной DREADD (дополнительный рис. 6м-х), что позволяет предположить, что вызванное стрессом повышение активности MeA препятствовало какой–либо дальнейшей реакции на их хемогенетическую стимуляцию. Химиогенетическая стимуляция нейронов MeAVMH не изменяла чувствительность к инсулину, но повышала уровень глюкозы в крови у мышей с гипогликемией, которым вводили инсулин для снижения исходного уровня глюкозы (дополнительный рис. 6l). Это говорит о том, что нейроны MeAVMH вряд ли участвуют в регуляции гомеостаза глюкозы в крови с помощью механизмов, включающих инсулин и другие классические глюкорегуляторные гормоны, но вместо этого способствуют адаптации гликемии, вызванной стрессом, с помощью неизвестного механизма. Хемогенетическая активация MeAVMH-нейронов не влияла на потребление пищи мышами натощак или на тревожное поведение (дополнительный рисунок 6e–g), что позволяет предположить, что MeAVMH-нейроны специфически вовлечены в гликемические реакции на стрессовые стимулы. Затем мы использовали оптогенетическую стимуляцию, чтобы оценить, может ли кратковременная активность в этом контуре вызвать изменения уровня глюкозы в крови, аналогичные тем, которые наблюдаются при длительной хемогенетической стимуляции. Оптогенетическая стимуляция MeAVMH в течение 5 мин и 15 мин значительно повышала уровень глюкозы в крови у мышей, экспрессирующих ChR2 без стресса, по сравнению с контрольными мышами, экспрессирующими eGFP (рис. 4q-s и дополнительный рис. 6z, aa), и повышала уровень глюкозы в крови после введения глюкозы (дополнительный рис. 6ab, ac). Аналогично результатам, полученным при химиогенетической стимуляции, вызванное стрессом повышение уровня глюкозы в крови не было дополнительно увеличено при оптогенетической стимуляции (дополнительный рис. 6ad–ak).
Чтобы оценить специфичность этих результатов на уровне circuits, мы охарактеризовали эффекты активации нейронов MeABNST. Мы обнаружили, что химиогенетическая стимуляция нейронов MeABNST не влияла на базальный уровень глюкозы или глюкозы в крови во время GTT (расширенные данные рис. 5n–p и дополнительные рис. 7c, d), уровни инсулина, глюкагона или кортикостерона в плазме крови (расширенные данные рис. 5b-p). 5q–t и дополнительный рис. 7e–g), пищевое поведение у мышей, которых кормили натощак (дополнительный рис. 7j), или поведение, похожее на беспокойство (дополнительный рис. 7h,i). Аналогичным образом, оптогенетическая стимуляция в течение 5 или 15 минут существенно не изменяла базовый уровень глюкозы, толерантность к глюкозе или реакцию на глюкозу в ответ на 5–минутное или 30-минутное ограничение или стрессовые факторы, связанные с территориальной клеткой (дополнительный рис. 7k-z). В совокупности эти данные позволяют предположить, что нейроны MeAVMH регулируют гипергликемическую, но не гипофагическую или поведенческую адаптацию к стрессу посредством механизма, независимого от основных гормонов глюкорегуляции надпочечников и поджелудочной железы.
A MeA–liver circuit regulates glucose
Предыдущие исследования подтвердили центральную регуляцию функции печени30,31. Таким образом, мы предположили, что нейроны MeA повышают уровень глюкозы в крови, "минуя" системы надпочечников и поджелудочной железы, и непосредственно стимулируя выработку глюкозы печенью. Такой механизм позволил бы нейронам MeA быстро повышать уровень глюкозы в крови во время стрессовых событий независимо от относительно медленно действующих гормонов глюкорегуляции. Чтобы исследовать эту возможность, мы сначала исследовали, взаимодействуют ли нейроны MeA с печенью посредством синаптических связей. Таким образом, мы ввели рекомбиназу Cre, экспрессирующую AAV1, в MeA мышей-репортеров Ai14 для антероградного и транссинаптического флуоресцентного мечения нижележащих нейронов32. У этих мышей нейроны в MeA и те, которые получают синаптическую информацию от MeA, экспрессируют красный флуоресцентный белок tdTomato (tdTom) в ответ на события рекомбинации, опосредованные Cre. Мы ввели вирус псевдорабиоза (PRV), экспрессирующий GFP (PRV-GFP), в печень этих же мышей (рис. 4f). Поскольку PRV-GFP ретроградно перемещается от места инъекции вдоль синаптически связанных нейронов, это позволило нам составить карту GFP+ нейронов, которые обеспечивают полисинаптическую связь с печенью (рис. 4f). Мы обнаружили в VMH клетки с двойной меткой, которые совместно экспрессировали как tdTom, так и GFP (рис. 4g). Это позволяет предположить, что нейроны MeAVMH взаимодействуют с печенью через сети полисинаптических связей. Мы предположили, что нейроны MeAVMH передают сигналы в печень через симпатические эфферентные нейроны вегетативной нервной системы. В соответствии с этой возможностью, опосредованная DREADD активация нейронов MeAVMH повышала экспрессию FOS в нейронах, экспрессирующих тирозингидроксилазу (TH) в лазурном месте (рис. 4h, i) и в TH-позитивных нейронах в ганглиях целиакии (рис. 4j-l), которые являются основными центральными и периферическими центрами, соответственно, симпатического эфферентного пути. Кроме того, активация нейронов MeAVMH увеличивала интенсивность TH в coeliac нейронах ганглиев (рис. 4м), что свидетельствует об активации симпатической нервной системы33.
Симпатическая активность регулирует выработку глюкозы в печени, модулируя синтез глюкозы de novo (глюконеогенез) и расщепление гликогена (гликогенолиз). Глюконеогенез основан на превращении пирувата в оксалоацетат и, в конечном счете, в глюкозу34. Таким образом, глюконеогенез можно оценить путем мониторинга повышения уровня глюкозы в крови после введения пирувата. В соответствии с регуляцией глюконеогенеза печени MeA, химиогенетическая стимуляция нейронов MeAVMH повышала уровень глюкозы в крови у мышей без стресса после введения пирувата (2 г/кг-1 внутрибрюшинная инъекция), как и химиогенетическая или оптогенетическая стимуляция нейронов MeA (рис. 4n, Расширенные данные рис. 6a, b и дополнительная рис. 8a–в). Затем мы оценили влияние модуляции MeAVMH на экспрессию глюкозо-6-фосфатазы (G6Pase, кодируемой G6pc (также известной как G6pc1)) и других чувствительных к стрессу генов печени, которые регулируют глюконеогенез (рис. 1d и расширенные данные рис. 1t). Активация нейронов MeAVMH у мышей, не подвергшихся стрессу, увеличивала экспрессию в печени Foxo1, активатора транскрипции глюконеогенных генов35 и G6pc, продукт которого контролирует конечную стадию глюконеогенеза в печени, ограничивающую скорость36 (рис. 4о), хотя мы не обнаружили каких-либо изменений в уровнях белка G6Pase у мышей, не подвергшихся стрессу, в этот момент времени (Рис. 4p и дополнительный рис. 8g,i–l). Стимуляция MeAVMH-нейронов у мышей без стресса увеличивала содержание гликогена в печени (расширенные данные, рис. 6г), что позволяет предположить, что вызванная стрессом активация MeAVMH-нейронов может увеличить общую способность печени вырабатывать глюкозу без распада гликогена. Затем мы оценили влияние хемогенетического подавления нейронов MeAVMH на экспрессию печеночных генов, которые регулируют глюконеогенез у мышей, подвергшихся стрессу. Мы обнаружили, что подавление активности нейронов MeAVMH не влияло на толерантность к пирувату, но притупляло вызванное стрессом повышение экспрессии гена G6pc в печени, увеличение экспрессии генов Irs2 и Igfbp1, чувствительных к инсулину в печени, и снижение уровней белка G6Pase в печени и белка PCK1 в печени, без влияния на содержание гликогена в печени (рис. 1). 6c–f и дополнительный рис. 8d,h–l).
Приведенные выше данные свидетельствуют о том, что нейроны MeAVMH регулируют гипергликемию, вызванную стрессом, с помощью механизма, включающего симпатическую регуляцию печеночного глюконеогенеза. Чтобы непосредственно проверить эту гипотезу, мы провели модифицированный тест на толерантность к пирувату с использованием [2,3-13C]пирувата (2 г/кг-1) у мышей, которых голодали в течение 4 ч в сочетании с оптогенетической стимуляцией нейронов MeAVMH. В соответствии с исследованиями хемогенетической модуляции, оптогенетическая стимуляция нейронов MeAVMH повышала уровень глюкозы в крови после введения пирувата (рис. 4т и дополнительный рис. 8е). Масс-спектрометрический анализ ткани печени, полученной через 15 мин после внутрибрюшинного введения [2,3-13С]пирувата (дополнительная фиг. 8f) подтвердили, что активация нейронов MeAVMH увеличивает включение 13C в ключевые глюконеогенные промежуточные продукты - цитрат M2, фумарат M2, аспартат M2, яблочную кислоту M2 и глутамат M2 (рис. 4u), а также среднее содержание 13C в этих промежуточных продуктах (расширенные данные рис. 6h). Меченый углерод также был включен в состав глицерин-3-фосфата M2, который может действовать как глюконеогенный субстрат. Примечательно, что оптогенетическая стимуляция нейронов MeAVMH увеличивала маркировку стабильными изотопами глюкозы, которая была оценена по первым двум атомам глюкозы во фрагменте глюкозы [1,2-13C], а по последним трем атомам глюкозы во фрагменте глюкозы [3-6-13C] (рис. 4u и расширенные данные на рис. 4). 6i; адаптировано по ссылке 37). В совокупности эти данные указывают на то, что активность MeAVMH-цепи усиливает глюконеогенез в печени, повышая уровень глюкозы в крови.
Repeated stress blunts the MeA–liver circuit
Наконец, мы исследовали, изменялась ли регуляция гликемии с помощью MeA в результате многократного воздействия стресса. Это важно, поскольку известно, что стресс приводит к метаболическим нарушениям, включая T2D и ожирение
38-40. Сначала мы изучили влияние повторяющегося острого стресса, связанного с территориализацией клеток, на гомеостаз глюкозы. После базового периода содержания в чистой клетке (5 мин) мышей подвергали воздействию специфического запаха в обжитой (территориализированной) клетке (2 мин) с последующим возвращением в знакомую чистую клетку (3 мин), причем этот цикл повторялся в общей сложности 5 раз в течение одного сеанса тестирования. Для каждого стрессового воздействия использовалась отдельная территориализированная клетка, чтобы предотвратить привыкание к стрессору. Как и ожидалось, первоначальное воздействие территориального стресса в клетке повышало уровень глюкозы в крови; однако последующие воздействия не оказывали никакого влияния на уровень глюкозы в крови (рис. 5d), хотя максимальный эффект мог предотвратить дальнейшее повышение. Это притупление вызванной стрессом гипергликемии отражалось на нейронной активности MeA, при этом первоначальное воздействие значительно увеличивало активность MeA, в то время как последующие воздействия были гораздо менее эффективными (расширенные данные на рис. 7a–c). Аналогичным образом, активность нейронов MeA
VMH была значительно повышена во время первоначального воздействия территориализированного клеточного стресса, но не при последующих воздействиях с использованием того же повторяющегося цикла (рис. 5a–c и расширенные данные рис. 7d). Нейроны MeA
VMH также продемонстрировали притупление активности в ответ на стресс в клетке, когда интервал между воздействиями был увеличен с 5 минут до каждых 6-12 часов (Расширенные данные на рис. 7e–h). Их активность также снижалась, когда мышей подвергали ограничительному стрессу дважды в день (30-минутные сеансы) в течение 5 дней, что сопровождалось ослаблением экспрессии MeA FOS и вызванной стрессом гипергликемией (рис. 5е,f). Эти данные свидетельствуют о том, что нейроны MeA
VMH становятся устойчиво невосприимчивыми к стрессу после первоначального воздействия.
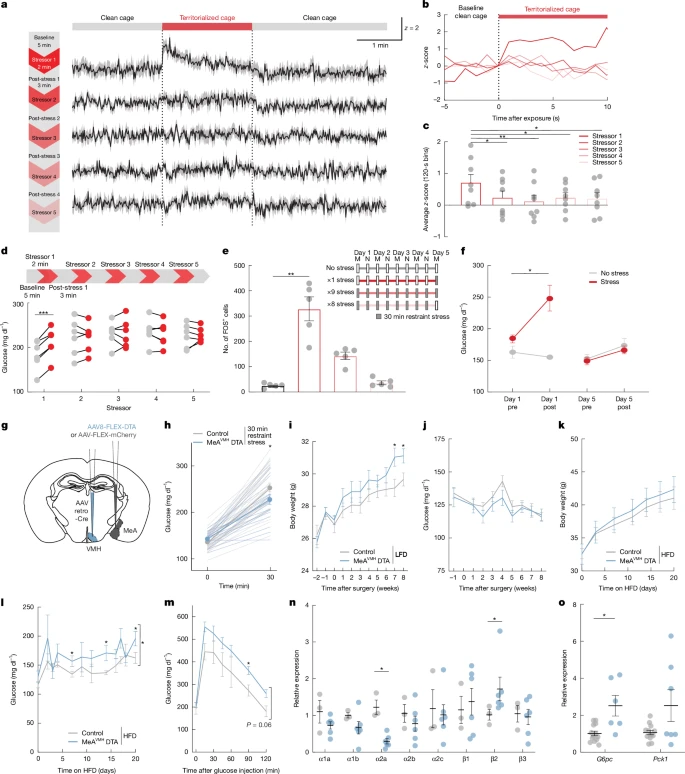 Рис. 5: Притупление MeA>Активация нейронов VMH при хроническом стрессе способствует увеличению веса и гипергликемии.
Рис. 5: Притупление MeA>Активация нейронов VMH при хроническом стрессе способствует увеличению веса и гипергликемии.
Мы исследовали функциональное значение подавляющего воздействия многократного стрессового воздействия на активность MeA. С этой целью мы двусторонне истощили MeAVMH-нейроны путем условной экспрессии субъединицы А дифтерийного токсина (DTA) в этих нейронах (рис. 5g), что привело к уменьшению количества MeA-нейронов на 30% (расширенные данные рис. 8a). Чувствительность к инсулину, а также толерантность к глюкозе и пирувату не отличались у мышей, подвергшихся воздействию MeAVMH-DTA, и у мышей контрольной группы (дополнительный рисунок. 9o–q). Аналогичным образом, уровни инсулина, глюкагона, кортикостерона, адреналина и норадреналина в плазме крови (расширенные данные, рис. 8с–г) и поведение в тесте в открытом поле (дополнительный рис. 9с, г) не отличались у мышей, подвергшихся MeAVMH-DTA, от контрольных мышей. Однако мыши, у которых не было MeAVMH-DTA, продемонстрировали притупленные гипергликемические реакции на ограничение (5 мин и 30 мин) и стрессовые ситуации в клетке (5 мин) (рис. 5h, Расширенные данные Рис. 8b и дополнительная фиг. 9e–n). Это согласуется с ослабленной гипергликемической реакцией на стресс, наблюдаемой у мышей после острого подавления MeAVMH-нейронов, опосредованного DREADD. Поскольку хронический стресс и выбор рациона питания тесно взаимосвязан41, мы изучили влияние истощения нейронов MeAVMH на рацион с высоким содержанием жиров. Мы не наблюдали различий в количестве нейронов FOS+ в лазурном луче или coeliac ганглиях у мышей, не получавших MeAVMH-DTA, по сравнению с контрольными мышами, получавшими диету с высоким содержанием жиров, что позволяет предположить, что базальная центральная и периферическая симпатическая активность у поврежденных мышей существенно не изменялась (дополнительный рисунок 9r, s). Симпатическая иннервация печени действует через a2A-адренорецепторы, подавляя индуцируемый глюкагоном цАМФ и выработку глюкозы42 и улучшая толерантность к глюкозе у мышей с ожирением43, в то время как α2-адренергическая сигнализация увеличивает выработку глюкозы печенью 44. Мы обнаружили, что у мышей с дефицитом MeAVMH-DTA снижалась экспрессия α2A-адренергических рецепторов и повышалась экспрессия β2-адренергических рецепторов в печени (рис. 5n), что, как ожидалось, должно было увеличить выработку глюкозы печенью. Соответственно, мы обнаружили, что мыши, пораженные MeAVMH-DTA, имели более высокие уровни глюкозы в крови и нарушенную толерантность к глюкозе по сравнению с контрольными мышами, когда им давали диету с высоким содержанием жиров (рис. 5l, m и дополнительный рис. 9b). Более того, экспрессия глюконеогенного гена G6pc в печени была значительно повышена у мышей, пораженных MeAVMH-DTA (рис. 5о). Мыши, пораженные MeAVMH-DTA, получавшие стандартное питание, также имели увеличенную массу тела по сравнению с контрольными мышами, без изменений в потреблении пищи или уровнях глюкорегуляторных гормонов (рис. 5i,j и расширенные данные рис. 8c,d,h–j). Это влияние на массу тела еще больше не усиливалось, когда мышам давали диету с высоким содержанием жиров (рис. 5к и расширенные данные рис. 8к). Эти данные свидетельствуют о том, что периодически возникающий дефицит активности нейронов MeAVMH, вызванный стрессом, может повышать уязвимость к метаболическим нарушениям, включая увеличение веса и T2D-подобные нарушения уровня глюкозы в крови.
Резюме
Стресс вызывает высокоорганизованные метаболические реакции, которые играют решающую роль в поддержании поведенческой адаптации к стрессу, необходимой для выживания. Нейронные механизмы, лежащие в основе метаболической пластичности, связанной со стрессом, в значительной степени неизвестны. Наши результаты указывают на важную роль популяции нейронов MeA, отвечающих за гипоталамус, в регуляции гипергликемических реакций на физические и социальные стрессоры. Неожиданно оказалось, что нейроны MeA регулируют уровень глюкозы в крови независимо от классических гормонов глюкорегуляции надпочечников и поджелудочной железы. Мы обнаружили, что нейроны MeA обеспечивают полисинаптическую связь с печенью через симпатическую нервную систему, стимулируя глюконеогенез в печени. Наши результаты убедительно доказывают, что миндалевидные системы управляют метаболическими реакциями на стресс посредством быстрого высвобождения глюкозы печенью. Многократное воздействие стресса вызывало адаптацию активности этих гепаторегуляторных нейронов MeA, что приводило к постоянному повышению уровня глюкозы в крови. Если эти результаты распространяются на людей, то они предполагают, что нарушение регуляции передачи сигналов MeA способствует увеличению частоты метаболических нарушений у людей, подвергающихся длительным периодам стресса.
