Интересно, что аминокислота К650 (которая вызывает TD-II при мутации в Е650) м. мутаировать двумя дополнительными способами, приводя к двум разным заболеваниям. При мутировании в М650 (найдена у 4 неродственных индивидов) она вызывает SADDAN. SADDAN пациенты имеют скелетную дисплазию и доживают до перинатального периода. Помимо этого acanthosis nigricans и структурных аномалий ЦНС и функциональных нейрологических нарушений, такиех как потеря слуха, наблюдаеются у выживших пациентов с SADDAN. Если мутация в N650, то это приводит к НСН, синдрому с короткими конечностями, но с более слабой скелетной дисплазией, чем при АСН, TD, SADDAN. HCH м. также возникать в результате ряда мутаций в др. доменах FGFR3 (Табл.).
Сходство скелетных дисплазий с градированной тяжестью при НСН, АСН, TD и SADDAN указывает на общую основу этих заболеваний. Имеющиеся данные указывают на то, что градированная тяжесть является скорее количественным признаком, чем качественным. Во-первых. пациенты, гетерозиготные по АСН и НСН аллелям имеют более строгий фенотип, чем те, у которых затронут только один аллель. Во-вторых, пациенты, гомозиготные по АСН аллелям имеют фенотипы, сходные с таковым у TD пациентов. В-третьих, если К644Е мутация (которая соответствует К650Е) у людей) вносилась в Fgfr3 мышей, то она экспрессировалась на 10% от уровня дикого типа Fgfr3. Гомозиготные мыши, несущие этот заметно ослабленный TDII аллель, обладают свойствами, напоминающими АСН, тогда как гетерозиготные мыши обладают слабой скелетной дивсплазией, мимикрирующей НСН. Однако, если та же самая мутация экспрессируется на уровне, эквивалентном аллелю дикого типа. то вызываемый фенотип воспроизводит TD. Анализ in vitro показал, что FGFR3 кДНК, несущая мутации по АСН и TD, обнаруживает градированную активацию их тирозин киназных активностей (дикий тип ←ACH←TD).
Итак, получены десятки животных моделей, несущих различные мутации Fgfr3 (Табл. 3). В разной степени эти мыши воспроизводят соответств. болезни к людей (Brode and Deng, 2003). Изучение этих мышей показало. что, сигналы FGFr3 играют важную роль в ингибировании пролиферации и дифференцировки хондроцитов. Следовательно, назависимя от лиганда активация Fgfr3 вызывает значительное снижение хондрогенеза и роста кости, ведущее к карликовости. Напротив, мутантные мыши, несущие Fgfr3-нулевые мутации обнаруживают повышенную пролиферацию хондроцитов в пролиферативной зоне ростовой пластинки, приводя к быстрому и продолжительному эндохондральному росту костей. Это наблюдщение ведёт к заключению, что FGFR3 является негативным регулятором роста кости. Дальнейший анализ выявил участие многих нижестоящих молекул в трансдукции множественных сигналов, в том числе и Stats, ингибторов клеточного цикла и PTHrP.
Анализ таких мышей проводился в основном в перинатальном или раннем натальном возрасте. Несмотря на это был проведен детальный анализ с использованием моделей по мутациям TDII, Fgfr3-K644E, при которых мутантные мыши погибали при рождении. Интересно, что мутантные эмбрионы обнаруживали повышенной пролиферацией хондроцитов ростовых пластинок во время ограниченного времени на ранних ст. эндохондральной оссификации (Е14-15). Однако на ст. Е18 мутантные хондроциты пролиферировали со скоростями, сходными в контроле. Напротив, пониженная дифференцировка хондроцитов продолжалась в течение всего исследуемого периода развития диннх костей. Значение этого наблюдения в том, что только супрессия дифференцировки во время эбриональных стадий развития достаточна для для задержки эндохондрального роста кости, тогда как эффективное подавление роста длинных костей на постнатальных стадиях нуждается в нарушении и прролиферации и дифференцировки. Это наблюдаение говорит также против того, что причина укорочения конечностей у моделей TD м.б. отличной от причин у др. моделей активации Fgfr3, у которых фенотип становится очевидным на постнатальных стадиях.
Cancer
Мутации, которые активируют FGF рецепторы, чаще всего FGFR3, обнаруживаются также во многих формах рака. Так, описаны две мутации в FGFR2: S267P в IgIIIa и мутация сплайс-сайта (940-2A→G) в IgIIIc, которые найдены у пациентов с раком желудка. Интересно. что эти гетерозиготные соматические мутации идентичнызародышевым активирующим мутациям, которые ответственны за CS, AS и PS. Активирующие мутации FGFR3, которые были идентифицированы ранее при летальных и нелетальных скелетных дисплазиях, обнаружены и при раке мочевого пузыря. Скрининг FGFR3 мутаций при колоректальной карциноме у людей выявил новые мутантные транскрипты с аберрантным сплпайсингом и активацией скрытых сплайс-последовательностей с высокой частотой, до 50% у 36 с первичными опуходями и до 60% в 10 линиях клеток и колоректальных раков. Дополнительные миссенс мутации FGFR3 найдены в цервикальных карциномах и уротелиальных папиломах.
Крометого, хромосомная транслокация, которая ведет к активации FGFR1 или FGFR3 описана при ряде опухолей. Транслокация локуса тяжелой цепи иммуноглобублина в хромосому 14q32 вблизи FGFR3 является общераспространённым процессом при миэломах. FGFR3 экспрессируется на высоких уровнях в этих же опухолях. Некоторые из этих раковых опухолей несут также соматические мутации, Y373C, K650E и К650М, которые ответственны за скелетные дисплазии коротких конечностей. Хромосомные транслокации, затрагивающие FGFR1 описаны при клинических синдроме stem-cell myeloproliferative disorder (B-cell or T-cell lymphoblastic leukemia/lymphoma witj myeloid hyperplasia and peripheral blood eosinophilia). Предположительно слитые белки, FOP-FGFR1, ECP1-FGFR1, и FIM-FGFR1, являются онкогенными благодаря конституитивно активной киназе, запускаемой димеризацией с помощью межбелковых взаимодействий мотивов FGFR1. Исследование с помощью культуры клеток Ba/F3 показало, что FOP-FGFR1 индуцирует жизнеспособность клеток, опосредуемую с помощью МАРК и PI 3-киназы/Akt/mTOR пути.
 Scheme of the interaction between FGFR and primary components of Ras/MAP kinase pathway
Scheme of the interaction between FGFR and primary components of Ras/MAP kinase pathway
Signalling Through FGFR
Membrane Events Linked to Activation of FGFRs
FGF рецепторы обычно находтся в виде неактивных мономеров, они активируются после связывания лигнада посредством классического многоступенчатого пути. Теоретически две молекулы FGF (и гепарин) соединяются с внеклеочными частями IgII и IgIII рецептора, это ведет к гомодимеризации. Кристаллическая структура FGF2 и FGFR1 показывает важность остатка Цис для образования стабильного комплекса. IgII взаимодействует с лигандом через гидрофобные взаимодействия, тогда как IgIII взаимодействует через полярные и гидрофобные взаимодействия (Stauber et al., 2000). Димеризация сводит вместе внутриклеточные домены рецепторов, приводя к аутофосфорилированию некоторых критических остатков тирозина. Это в свою очередь позволяет связать FRS2 (GFGR stimulated 2 Grb2 binding protein) посредством SH2 (Src homology 2) домена и последующего привлечения Grb2 (growth factor receptor-bound protein 2), Sos (son of sevwnless nucleotide exchange factor), SHP2 (Src homology 2 phosphatase 2) и Shc. Эти заключенные в мембрану комплексы делают возможной активацию Ras, Raf, PLCγ1 и передачу сигналов MEK/ERK. Активация различных протеин киназ или транскрипционных факторов, таких как PLCγ1, PI3K, STAT, Сиаф1б Sox9, Src и т.д., описана в (Liu et al., 1999; Ong et al., 2001; Xu and Goldfarb, 2001).
The MAPK, PLCγ1 and PI3K Pathways
Ряд различных систем указывает на участии MAPK, PLCγ1 and PI3K в активации передачи сигналов FGF. У эмбрионов мыши показано, что известные или предполагаемые области пердачи сигналов FGFR перекрываются ( но не полностью покрывают) домены наиболее сильной активации ERK. Кратковременная инкубация с ингибитором FGF рецептора специфически снижает phospho-ERK окрашивание.
Получены также доказательства активации PI3K и PLCγ1 путей; напр., стимуляция с помощью FGF2-FGFR1 клеток adrenal cortex capillary endothelial (ACE) активирует МАРК и PI3K пути с помощью ERK и Akt, соотв. Используя гладкомышечные клетки (SMCs) было установлено. что FGF2 стимулирует р38 МАРК-ERK путь, который участвует в дифференцировке этих клеток, хотя не наблюдалось активации передачи сигналов PI3K.
Установлено, что воздействие FGF2 индуцирует эпителиальные клетки роговицы подвергаться EMT? что характеризуется пролиферацией и изменением формы клеток. Используя специфические ингибиторы, оказалось возможным связать PI3K путь с обоими событиями, а PLCγ1 путь только с активацией клеточных митозов. Подобно PI3K действию на Ras, белок PLCγ1 также важен и для активации FGF1-FGFR1-ERK2/ Эти исследования представили дополнтельные доказательства того, что отдельные FGF-активируемые пути м. действовать совместно или независимо.
Др. исследования проводились с участием МАРК и PI3K путей в ингибировании апоптоза или выживаемости. Было установлено, что воздействие all-trans ретиноевой кислоты на клетки эмбриональной карциномы Р19 вызывает активацию каспазы 3, ведущей к апоптозу. Присутствие FGF2 ингибирует 90% каспаза-3-подобной активности благодаря усилению фосфорилирования Bad, анти-апоптического члена семейства Bcl-2. Эти события были непосредственно ассоциированы с активацией PI3K. Недавно было усатновлено, что жизнеспособность дофаминергических нейронов, чья дегенерация связана с проявлениями болезни Паркинсона, увеличивается благодаря стимуляции Fgfr1c и МАРК с помощью FGF20.
Передача сигналов FGF участвует также в процессах дифференцировки. Установлено. что Fgfr2, действующий посредством пути PI3K/PKC/Act, необходим для дифференцировки ES клеток. Доминантная мутантная форма Fgfr2 супрессирует экспрессию некоторых белков внеклеточного матрикса ( laminin a1, b1, g1; collagen IV a1 и а2). Предполагается, что передача сигналов FGF через Fgfr2/PI3K индуцирует экспрессию основных компонентов мембран, процесс участвующий в дифференцировке эпителия.
Недавно идентифицирован ряд ингибиоров петли обратной связи передачи сигналов FGFR, включая Sprouty, Sef и Pyst белки. Эти белки специфически ингибируют МАРК путь. Sef взаимодействует с Fgfr1, ингибируя фосфорилирование, связанное с активацией рецепторов. Sprouty2 фосфорилируется по тирозину после стимуляции с помощью FGFR или EGF, феномен, который увеличивает сродство к с-Cbl, нижестоящему регулятору рецептор-тирозин киназного пути. Наконец, Pyst1 экспрессия увеличивается с помощью предачи сигналов FGF (в нейральной пластинке цыплят) , а в результате негативной петли обратной связи активность Pyst1 снижает уровни phospho-MAPK.
Cbfa1 (Core-Binding Transcription Factor Alpha Subunit Type 1)
YtlоНедавно показано, что активация FGFR2 с помощью FGF2 или FGF4 стимулирует экспрессию и активацию Cbfa1 и что этот процесс скорее всего обеспечивается МАРК и РКС сигнальными путями. Cbfa1 является транскрипционным фактором, который контролирует дифференировку гипертрофических хондроцитов и остеобластов. Мыши, лишенные Cbfa1 не имеют остеобластов и обнаруживают дефицит в созревании хондроцитов. Пациенты. гетерозиготные по мутациям или делециям СИАФ1 обнаруживают cleidocranial dysplasia.
Показано, что in vivo передача сигналов FGF/FGFR действует выше Cbfa1. Упомянутые ранее Fgfr1-P250R мутанные мыши обнаруживают признаки, воспроизводящие таковые у CS пациентов, включая преждевременное слияние и коронарного и сагитального швов. Установлено, что мутантные остеобласты экспрессируют Cbfa1 на более высоком уровне, чем клетки дикого типа. Это наблюдение указывает на то, что Cbfa1 м.б. нижестоящей мишенью для сигналов Fgf/Fgfr1. Согласуется с этим и трансфекция С3Н10Е1/2 клеток дикого типа или мутантной Fgfr1 кДНК или обработка этих клеток Fgf2 или Fgf8, которые индуцируют экспрессию Cbfa1. Показано, что увеличение Cbfa1 постепенно активирует свои нижестоящие транскрипционные мишени, включая osteocalcin и bone sialoprotein, ведущие к ускорению дифференцировки остеобластов. Напротив. зедерзка оссификации коррелирует со снижением экспрессии Cbfa1 у эмбионов Fgfr2c-/-. Однако не обнаруживается видимых изменений в экспрессии Cbfa1, jsteocalcin и bone sialoprotein у мутантных мышей. несущих мутацию Fgfr2-S250W, которая соответствует у людей AS, это указывает на разные механизмы, лежащие в основе этих синдромов.
Signal Transduction and Activator of Transcription Proteins
Имеются указания на то, что активирующие мутации FGFR3 вызывают скелетные дисплазии, частично, действуя путём активации белков signal transduction and activator of transcription (STAT). Впервые продемонстрировано, что экспрессия FGFR3-K650E в культуре клеток активирует STAT1, который в свою очередь усиливает активность р21, регулятора клеточного цикла. Согласуется с этим и то, что хондроциты ростовой пластинки у пациентов с TDII обнаруживают высокие уровни активированного STAT1 и р21. Прелдполагается, что STAT1 v/ дейстовать как медиатор задержки роста в развитии кости путём регуляции ингибитороа клеточного цикла. Активация STAT белков наблюдалась также у большинства животных моделей карликовости с короткими конечностями. Трансгенные мыши с избыточной экспрессией FGF обнаруживают, что в отсутствие STAT1 они м. преодолевать ингибирование FGF сигналов на пролиферацию хондроцитов в культивируемых костях и у взрослых животных.
Анализ животных моделей выявил также, что активирующие мутации Fgfr3 вызывают активацию множественных Stats, включая Stat1, Stat5a и 5b, указывая тем самым на широкое влияние сигналов FGF на белки STAT. Фосфорилирование множественных белков STAT также вызывает экспрессию др. рецепторов, возможно посредством механизма, связанного с факторами активирующими тромбоциты, JAK-2 и Src. Исследования животных моделей также выявили повышенную экспрессию некоторых ингибторов клеточного цикла, включая р21, а также р16, р18 и р19, которые принадлежат семейству Ink4. Это указывает на тоя.что активированны STAT сигнал м. генерировать плейотропные эффекты, которые м.б. опосредованы множественными ингибиторами клеточного цикла и др. ещё не установленными факторами. В согласии с эти и то, что введение р21-нулевого фона мутантным мышам не оказывает влияния на скелетный фенотип возможно из-за перекрывания дрю ингибторами клеточного цикла..
Interaction between FGF/FGFR and IHH/PTHrP Signaling Pathways
Indian hedgehog (IHH)/parathyroid hormone-related protein peptide (PTHrP или PTHIP) сигналы представляют собой важный путь для роста костей. IHH и PTHrP функционируют в негативной петле обратной связи, контролируя пролиферацию и дифференцировку хондроцитов ростовой пластинки. Согласно этой модели IHH, который экспрессируется в хондроцитах зоны созревания, индуцирует экспрессию PTHrP в околосуставной надхрящнице. Активация PTHrP-R в прегипертрофических хондроцитах предупреждает их от дифференцировки в гипертрофические клетки.
Проверка мышиных моделей, несущих активированный Fgfr3 выявила заметно сниженную экспрессию IHH и PTHrP рецепторов. В культивируемых костных рудиментах, обработанных FGF2, продемонстрировано, что подавление IHH и PTHrP рецепторов происходит до появления костных аномалий. Это указывает на то, что FGFR3 действует выше IHH и PTHrP рецепторов и негативно регулирует их экспрессию. Учитывая позитивную роль сигналов IHH в пролиферации хондроцитов, это м. указывать на то, что подавление IHH вносит вклад в патогенез скелетных дисплазий, связанных с FGFR3. Мыши, дефицитные по IHH или PTHrP или PTHrP-R обладают преждевременно гипертрофированными хондроцитами, что характеризуется расширением зоны гипертрофических хондроцитов. Однако мыши, несущие активированный Fgfr3, несмотря на подавление IHH и PTHrP-R не обнаруживают каких-либо признаков преждевременной гипертрофии хондроцитов; вместо этого они обнаруживают заметное снижение размеров гипертрофических хондроцитов и сильное сужение зоны гипертрофических хондроцитов. Следовательно,передача сигналов FGF/FGFR3 ингибирует дифференцировку хондроцитов независимо от передачи сигналов IHH и PTHrP.
Установлено, что воздействие PTHrP м. ингибировать дифференцировку хондроцитов во всех культивируемых костях, независимо от их генотипа. Ита, эти наблюдаенияы и указывают, что FGF/FGFR3 и IHH/PTHrP сигналы ингибируют дифференцировку хондроцитов доминантным и независимым образом.
Изучение культур костей, выделенных от трансгенных мышей, несущих FGFR3-G380R, выявило сходную, но отдельную модель взаимодействий между FGF/FGFR, IHH/PTHrP и DVH/ N/r/ эта модель подтвержадет негативную роль передачи сигналов FGF в пролиферации хондроцитов, то предполагается, что предача сигналов FGF/FGFR ускоряет скорее, чем ингибирует гипертрофическую диффйеренцировку независим от системы IHH/PTHrP. Было предположено, что уменьшение гипертрофической зоны является вторичным по отношению к быстрой дифференцировке и медленной пролиферации хондроцитов. Хотя это объяснение согласуется с данными, полученными на культивируемых костях, однако это не согласуется с тем фактом, что Fgfr3-/- мыши обнаруживают ускоренную дифференцировку хондроцитов и увеличенные размеры гипертрофических хондроцитов. Эта модель также неспособна объяснить, почему быстрая дифференцировка д. вызывать даметную редукцию размеров гипертрофических хондроцитов у мышей, несущих knock-in мутации. которые активируют Fgfr3. Всё это указывает на то, что передача сигналов FGF и DVH действуют антогонистически во время хондрогенеза. В соответствии с этим воздействие на культивируемые кости. выделенные от FGHFR3-G380R мышей, DVH l/ устранять нониженную скоровсть пролиферации хондроцитов и уменьшенные размеры гипертрофической зоны. Это наблюдение наводит на мысль о терапии таким образом скелетных дисплазий.
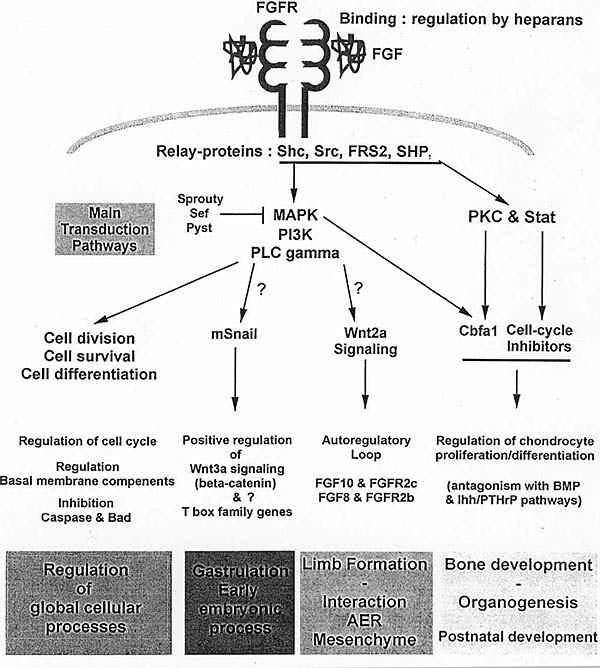 Рис. 4. A summary of the FGFR signaling and regulatory network discussed in the article
Рис. 4. A summary of the FGFR signaling and regulatory network discussed in the article

