Во время апоптоза секреция затухает (M Lowe M, VJ Allan et al., personal communication) и клетка обязательно разбирается по мембран-связанным blebs. Происходят сильные морфологические изменения секреторного пути, т.к. клетка демонтируется. ER разбухает и vesiculated [8]; однако, специфические события, вызываемые разборкой ER неизвестны.
Во время апоптической разборки комплекса Гольджи индивидуальные стеки (stacks) из цистерновых мембран теряют своё juxtanuclear положение и распадаются на пузырьки и тубулярные кластеры, сходно с митотической разборкой [8]. Некоторые белки с предполагаемой ролью в структуре Гольджи и трафике мембран, как недавно было установлено, являются субстратами каспаз, включая golgin-160 [9], GRASP65 (Golgi reassembly and stacking protein 65)
[10], giantin ([11] M Lowe M, VJ Allan et al., personal communication), p115 [12] и GM-130 ([11]; M Lowe M, VJ Allan et al., personal communication). Фактор t-SNARE (target membrane soluble N-ethylmaleimide sensitive factor attachment protein receptor) syntaxin-5 (M Lowe M, VJ Allan et al., personal communication) и два компонента dynein-dynactin моторного комплекса [13],особенно промежуточная цепь dynein и p150Glued субъединица dynactin, также являются субстратами каспаз во время апоптоза. Расщепление этих белков м. вносить вклад в арест мембранного трафика и способствовать воздействию липидов и углеводов, которые служат в качестве сигналов для фагоцитоза на поверхности клетки (M Lowe M, VJ Allan et al., personal communication). Расщепление структурных белков Гольджи необходимо для эффективной разборки Гольджи во время апоптоза: экспрессия не-расщепляющих мутантов GRASP65, p115 или golgin-160
задерживает апоптическую разборку Гольджи [9,12,13]. Интересно, что клетки, экспрессирующие non-cleavable golgin-160 демонстрируют задержку апоптоза, индуцированного с помощью стресса секреторного пути (CE Machamer et al., unpublished data), подтверждая тем самым, что golgin-160 расщепление м.б. вовлечено в передачу сигналов апоптической прогрессии также как и необходимо для эффективной разборки Гольджи. Плазматическая мембрана также разбирается во время апоптоза, т.к. мембран-связанные blebs, содержащие клеточное содержимое, выщипываются (pinch off) из клетки. Этот процесс в основном регулируется с помощью фосфорилирования легкой цепи миозина [14] Расщепление каспазой Rho-зависимой киназы ROCK I увеличивает её киназную активность, увеличивает фосфорилирование лёгкой цепи миозина и индуцирует blebbing мембраны на клеточной поверхности [15,16]. Ингибирование активности ROCK I блокирует blebbing плазматической мембраны во время апоптоза; однако, др. апоптические события остаются незатронутыми, включая фагоцитоз, воздействие phosphatidylserine и удаление сиаловой кислоты с клеточной поверхности [17], тем самым подтверждая, что blebbing плазматической мембраны независим от др. апоптических событий.
Stress responses in the secretory pathway
What induces secretory pathway stress?
Имеется несколько физиологически пригодных индукторов ER стрессов, включая накопление несвёрнутых или неправильно свёрнутых белков, исчерпание хранилищ кальция в просвете и продукцию β-amyloid [18]. Хотя не существует хорошо охарактеризованных Golgi-специфических стрессов, структура Golgi мембран м.б. особенно чувствительна к изменениям липидного состава. Напр., увеличение церамидов (ceramide) на комплексе Гольджи нарушает мембранный trafficking [19]. Др. потенциальные индукторы Golgi стрессов скорее всего включают сборку и почкование вирусов на Golgi или нарушения pH или содержания катионов. Обширный перенос (trafficking) между ER и Golgi м. иметь целью интеграцию стрессовых сигналов, ощущаемых в этих компартментах.
Endoplasmic reticulum stress can lead to repair
Unfolded protein response (UPR) в ER хорошо изучена. В клетках млекопитающих основными игроками являются два трансмембранных сенсорных белка, ATF6 и протеин киназа R-like ER kinase PERK [20]. Когда неупакованные белки накапливаются в ER, они смещают chaperone белок BiP/GRP78 из ATF6, который затем транспортируется в Гольджи, где он расщепляется с помощью тех же самых протеаз, которые высвобождают sterol-регулируемые транскрипционные факторы из мембраны [21,22]. Цитоплпазматическая часть ATF6 затем транслоцируется в ядро и индуцирует экспрессию chaperone белков, включая BiP/GRP78 и GRP94. Активация PERK также индуцирует фосфорилирование фактора инициирующего трансляцию eIF2a и глобальное подавление белкового синтеза. Это уменьшает груз белков в ER, давая накапливающимся белкам шанс сложиться и тем самым вывести клетки из стресса. Мутантные белки, которые не могут правильно сложиться обычно транслоцируются в цитоплазму для деградации с помощью протеасом [23]; Продолжительная UPR реакция которая не м.б. репарирована ведет к апоптозу. ER играет также центральную роль в гомеостазе кальция, служа в качестве большого резервуара для свободных ионов Ca
2+, которые соединяются с calcium-buffering белками в просвете ER. Свободный Ca
2+ накачивается через мембрану ER с помощью Ca
2+-зависимых ATPases, включая насос SERCA. Разные стрессы, включая оксидативные стрессы [24], запускают высвобождение Ca
2+ из ER. Calnexin и calreticulin являются calcium-связывающими haperones в ER, которые проверяют белки, как они уложены, а снижение уровней кальция в ER нарушает функцию calnexin и calreticulin, что вносит вклад в UPR [25].
Apoptotic signaling at the endoplasmic reticulum leads
to cell death
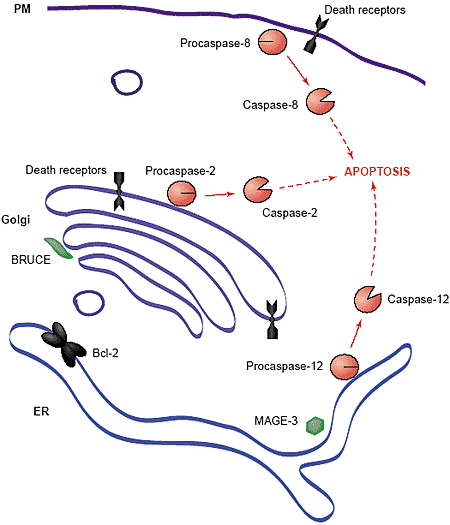
Caspase-12, обнаруживается на цитоплазматической поверхности ER
[26], она важна для индукции апоптоза в ответ на продолжительный ER стресс у мышей. Механизм активации caspase-12 неизвестен, но его выяснение м. прояснить, почему клетки в конечном итоге отказываются от репарации и инициируют апоптоз. Напр., д.б. procaspase-12 связывающие белки в ER, которые обеспечивают олигомеризацию и аутоактивацию procaspase-12 в ответ на ER стресс. Одним из таких кандидатов является TNFR-ассоциированный белок TRAF2, который соединяется с и способствует образованию кластеров procaspase-12 в клетках с избыточной экспрессией обоих белков [27]. Procaspase-12 м.б. расщеплена с помощью самой себя или с помощью calpain [26] и caspase-7 [28]. Возможно также, что ER стресс разрушает взаимодействие между procaspase-12 и ингибитором, таким как MAGE-3 (melanoma-associated antigen-3), который блокирует протеолитическую активацию procaspase-12
in vitro [29]. Стабильная экспрессия MAGE-3 делает клетки мышей более резистентными к ER-стрессами индуцированному апоптозу [29].
Хотя caspase-12, по-видимому, является критической для активации апоптоза в ответ на ER стресс у мышей, не идентифицировано у людей ортологи caspase-12.
Человеческие кДНК предположительно кодирующие caspse-12 имеют и преждевременный стоп кодон во всех сплайс-вариантах и дополнительно мутацию потери функции в законсервированной области, существенной для каспазной активности [30]. В отсутствие caspase-12, , следовательно, люди м. иметь альтернативный механизм активации апоптоза в ответ на ER стресс. Напр., каспаза с перекрывающейся функцией м. компенсировать отсутствие caspase-12. Одним из привлекательных кандидатов является caspase-2.
Помимо активации caspase-12, стресс м. вызывать высвобождение кальция, истощение ER хранилищ кальция. Thapsigargin необратимо ингибирует насос SERCA и вызывает истощение хранилищ кальция в ER. Клетки, обработанные thapsigargin усиливают экспрессию DR5, которые м. активировать передачу апоптических сигналов в ответ на родственный TNF апоптоз-индуцирующий лиганд [31], указывая на связь между истощением хрнаилищ кальция в ER и апоптической кухней. Исчерпание хранилищ кальция ER м. индуцировать апоптоз, возможно за счёт увеличения уровня митохондриального кальция. Анти-апоптический белок Bcl-2 частично локализуется в ER мембранах и м. регулировать гомеостаз кальция в ER.
Описаны разные эффекты Bcl-2, однако, и это наиболее вероятно, что тип и контекст клеток м. влиять на исход активности Bcl-2 [4]. Интересно, что обработка клеток с помощью TNF-α, который активирует DR TNFR-1, также активирует высвобождение ER кальция; этот эффект блокируется избыточной экспрессией Bcl-2 [32]. Механизм, с помощью которого внеклеточный лиганд вызывает высвобождение кальция из ER неизвестен, но он м.б. связан с DR trafficking и/или с acid sphingomyelinase-индуцированными изменениями клеточного липидного метаболизма.
Golgi stress and apoptotic signaling
Комплекс Гольджи, по-видимому, играет важную роль в апоптозе. Caspase-2 локализуется в комплексе Гольджи и ядре [9,33]. Т.к. нулевые по caspase-2 мыши жизнеспособны и обнаруживает только минимальные дефекты развития [34], но недавние эксперименты предоставили доказательства того, что caspase-2 играет центральную роль в передаче апоптических сигналов ([35]; CE Machamer et al., unpublished data). Caspase-2-дефицитные фибробласты мышей резистентны к апоптозу, вызываемому про-апоптическими лекарствами, которые индуцируют или UPR или др. секреторные стрессы, а экспрессия нерасщепляемого golgin-160 (субстрат для Golgi-localized caspase-2) ослабляет апоптоз в клетках человека, индуцируемый теми же самыми стимулами (CE Machamer et al., unpublished data). Потребность в Golgi-localized caspase-2 м. указывать на то, что UPR-индуцируемые стимулы затрагивают Гольджи в дополнение к ER, или что непрерывный перенос между ER и Golgi позволяет интегрировать передачу апоптических сигналов от этих компартментов.
Для capsase-12 механизм активации caspase-2 в комплексе Гольджи неизвестен. Единственным потенциальным негативным регулятором активации caspase-2 является baculoviral IAP-repeat-containing ubiquitin-conjugating enzyme (BRUCE), который локализуется в комплексе Гольджи [36]. Гомолог у Drosophila BRUCE м. супрессировать клеточную гибель с помощью про-апоптических белков Reaper и Grim [37], подтверждая тем самым, что он является IAP. Продукты расщепления каспазой белков, находящихся в Гольджи, м. иметь потенциальную роль в индукции апоптотоза. Конструкции, которые воспроизводят фрагменты каспазного расщепления golgin-160 и p115 локализуются в ядре [12,38], где они обнаруживают потенциал сигналов транскрипционной активации репарационной или апоптической кухни. Апоптическое расщепление и релокализация белков из Гольджи м. обеспечивать отсутствующую связь между стрессом в Гольджи и клеточной реакцией, аналогично UPR активации в ответ на расщепление ATF6.
DRs демонстрируют др. потенциальную связь между Гольджи и индукцией апоптоза. Большая часть внимания исследователей была обращена на DRs, присутствующие на плазматической мембране. Интересно, что огромное количество этих же самых DRs локализуется на комплексе Гольджи [39,40]. Перенос DR Fas на плазматическую мембрану стимулируется с помощью GD3 [41], ganglioside, продуцируемого в Гольджи, который участвует в качестве важной апоптической сигнальной молекулы [42]. Более того, локализованная в Гольджи caspase-2, по-видимому, необходима для апоптоза, сопровождающего ligation DRs TNFR-1 и
Fas в фибробластах мышей (CE Machamer et al., unpublished data). Эти данные указывают на то, что, по крайней мере, в некоторых типах клеток, апоптическая реакция на death лиганды м. зависеть от Golgi-опосредованной регуляции мембранного трафика внутри клетки.
Conclusions
Передача сигналов от мембран в секреторный путь позволяет клеткам ощущать специфические типы стрессов. Коммуникации между секреторными компартментами м. интегрировать стрессовые сигналы, делая возможной активацию специфических путей репарации и в конечном счёте инициируя апоптоз в ответ на нерепарируемые повреждения. Выяснение механизмов активации caspases-2 и -12 будет критическим для расшифровки передачи апоптических сигналов в секреторный путь. Понимание того, как стрессы ощущаются на секреторном пути мембранами и как эти сигналы достигают остальной части клетки, является следующей задачей выяснения пути клеточной гибели, затрагивающего секреторный путь.
Update
Потенциальная связь между ligation DRs и высвобождением ER кальция м. обеспечиваться с помощью трансмембранного ER резидентного белка BAP31. BAP31 расщепляется с помощью caspase-8 во время апоптоза, чтобы продуцировать интегральный мембранный p20 фрагмент и растворимый С-терминальный фрагмент [43].
Клетки, экспрессирующие caspase-резистентный BAP31 устойчивы к Fas ligation-индуцированному апоптозу [44]. Последняя работа показывает, что клетки, экспрессирующие p20 каспазой выщепленный фрагмент BAP31, подвергаются апоптозу и сенсибилизируются к caspase-8-индуцированному высвобождению
cytochrome c из митохондрий [45]. Этот процесс блокируется с помощью обработки calcium chelators или истощения хранилищ ER кальция до добавления апоптических стимулов, указывая тем самым. что кальцием-обусловленная передача сигналов между ER и митохондриями необходима для подобных коммуникаций
[45].
Сайт создан в системе
uCoz