Integrating transcriptional and signalling networks during
muscle development  Current Opinion in Genetics & Development 2004, 14:1–8 | |
|
A fundamental aspect of developmental decisions is the
ability of groups of cells to obtain the competence to respond to
different signalling inputs. This information is often integrated
with intrinsic transcriptional networks to produce diverse
developmental outcomes. Studies in Drosophila are starting
to reveal a detailed picture of the regulatory circuits controlling
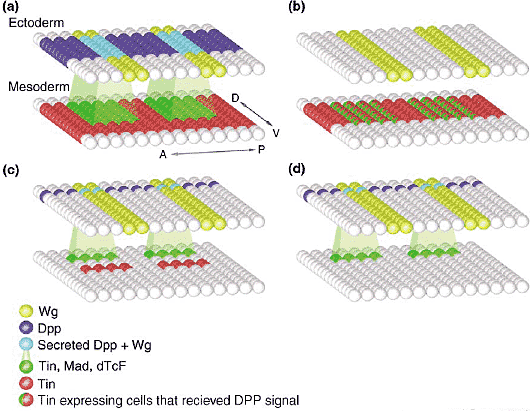
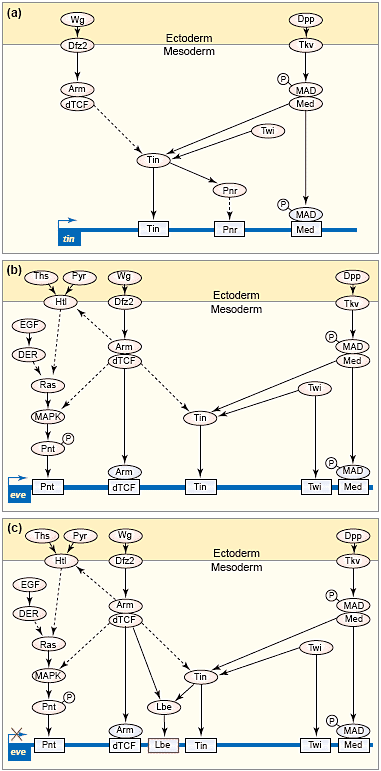
the subdivision of the dorsal mesoderm, which gives rise to
diverse muscle types including cardioblasts, pericardial cells,
body wall muscle and gut muscle. The combination of a common
set of mesoderm autonomous transcription factors (e.g. Tinman
and Twist) and spatially restricted inductive signals (e.g. Dpp
and Wg) subdivide the dorsal mesoderm into different
competence domains. The integration of additional signalling
inputs with localised repression within these competence
domains results in diverse transcriptional responses within
neighbouring cells, which in turn generates muscle diversity. | |