Myocardin открыт при скрининге новых генов, экспрессирующихся специфически в сердце [3]. Myocardin стимулирует активность SRF путем формирования четвертичного комплекса с SRF на ДНК и создает его строгий transcriptional activation domain (TAD) для SRF, который во всём остальном является очень слабым активатором транскрипции. Myocardin преимущественно активирует промоторы, содержащие два или более CArG боксов, это наблюдение согласуется с известной кооперативностью CArG боксов, ассоциированной с мышечными генами. Однако, имеются драматические отличия в чувствительности SRF-зависимых промоторов к myocardin, которые не м.б. объяснены просто количеством CArG боксов или их сродством к SRF. Вносят ли позиция и фланкирующие последовательности CArG боксов вклад в чувствительность к myocardin, предстоит вцяснить. Активация транскрипции с помощью myocardin также исключительно чувствительна к уровню SRF, так, что даже незначительное увеличение экспрессии SRF м. приводить к супрессии активности myocardin, возможно благодаря механизму 'squelching', с помощью которого SRF оттитровывает myocardin от SRF генов мишеней [3].
Идентифицированы два дополнительных члена семейства myocardin, обозначенных MRTF-A (MAL, MKL1, BSAC) и MRTF-B (MKL2) (Figure 1)
[3,4,27-29]. Ген, кодирующий MRTF-A (MAL/MKL1) человека оказывается транслоцированным при острой megakaryocytic лейкемии, при этом образуется онкогенный слитый белок с новым белком. названным One-twenty-two (OTT)/RBM15 (RNA binding protein motif protein 15) [27,28]. Слитый
RBM15-MKL1 белок обладает повышенной транскрипционной активностью в отношении индуцибельных ростовым фактором промоторов
c-fos и Egr1, но его способность активировать SM гены сравнима с белком MKL1 дикого типа [30]. Почему этот слитый белок обнаруживает повышенную активность в отношении growth-associated SRF генов мишеней неясно, но такая активность м. объяснять его лейкемогенные эффекты. Специфические гены-мишени из MRTF-SRF комплекса, ответственные за онкогенез, также не установлены. MRTF-A был также идентифицирован при скрининге генов, которые защищают от tumor necrosis фактора, индуцирующего клеточную гибель и названного
BSAC [29].
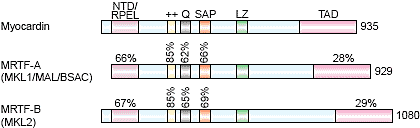 Рис. 1. Schematic diagrams of myocardin and MRTFs. Structural domains of myocardin and MRTFs are shown. ++, basic region; Q, glutamine-rich region. The percent identity between different domains is shown.
Рис. 1. Schematic diagrams of myocardin and MRTFs. Structural domains of myocardin and MRTFs are shown. ++, basic region; Q, glutamine-rich region. The percent identity between different domains is shown.
В то время как имеются три гена, кодирующих членов семейства myocardin, у млекопитающих, имеется, по-видимому, четыре таких гена у
Xenopus и puffer-fish (Fugu rubripes) (D Wang, EN Olson, unpublished; Krieg et al., personal communication). Одиночный родственный myocardin ген имеется у
Drosophila. Продукт этого гена, названный DMRTF, стимулирует активность
Drosophila SRF и необходим для формирования терминальных областей трахейной системы, примитивного органа по сравнению с ветвящимися трубками, участвующими в газообмене, которые функционируют аналогично сосудистой системе позвоночных (Z Han, EN Olson, unpublished). Это удивительно в отношении молекулярного партнерства myocardin и SRF , которое эволюционно законсервировано, чтобы регулировать развитие ветвящейся трубчатой сети, участвующей в обмене кислорода и питательных веществ.
Structural features of myocardin proteins
Myocardin принадлежит к семейству белков SAP (SAF-A/B, Acinus, PIAS), которое выполняет разнообразные роли в ремоделировании хроматина, транскрипционном контроле и фрагментации ДНК во время апоптоза [31]. Домен в 35 аминокислот SAP, как полагают, приобретает helix-linker-helix структуру с потенциалом связываться с ДНК. SAP домен у SAF-A связывается с A/T богатой геномной ДНК, ассоциированной с ядерным матриксом, т.наз. областью прикрепления ядерного матрикса. Сходным образом домен SAP в myocardin необходим для ассоциации myocardin с A/T богатолй ДНК (Z Wang, E Olson, unpublished). Интересно, что делеция SAP домена myocardin устраняет способность активировать ген
atrial natriuretic factor
(ANF), маркер кардиальной реакции на стрессы, не влияя на активацию гена SM22 [3]. Эта находка указывает на то, что домен SAP в myocardin м. различать между SRF генами мишенями, возможно благодаря ассоциации с дополнительными факторами.
Myocardin и MRTFs содержат консервативный N-терминальный домен (Рис. 1 и 2), состоящий из RPEL повторов, которые участвуют в Rho-зависимом ядерном импорте MRTF-A/MAL [32]. Делеция N-терминального домена myocardin или MRTFs усиливает транскрипционную активность белков. Базовый домен myocardin семейства белков способствует ядерной локализации, а также SRF взаимодействию [3], a leucine zipper-like домен способствует гомо- или гетеродимеризации между членами семейства myocardin [3,4, 32,33]. Димеризация м. обеспечивать механизм взаимосвязи миокардина или MRTF/SRF комплексов, связывающихся с разными CArG боксами внутри контрольных областей SM генов.
Транскрипционная активность myocardin обеспечиваетя с помощью
TAD на с конце белка [3]. Даже вся аминокислотная последовательность TADs только на 30% идентична у myocardin и MRTFs, они функционально взаимозаменяемы (S Li, Z Wang, EN Olson, unpublished). TADs у myocardin и MRTFs м.б. также замещена VP16 TAD, показывая тем самым, что этот домен не обладает специфичностью в отношении активации генов мишеней [3]. Установлено, что TAD у myocardin ассоциирует с транскрипционным ко-активатором p300, гистоновой ацетил-трансферазой, это открывает возможность, что myocardin м. управлять хроматиновой структурой SRF генов мишеней, ключевой ступенью в активации программ дифференцировки SM [34,35].
Expression patterns of myocardin and
MRTFs
Во время эмбриогенеза мышей экспрессия myocardin впервые выявляется в кардиальных клетках предшественниках внутри кардиального серпа на ~E7.5 и после этого поддерживается в кардиальных миоцитах атриальных и вентрикулярных камер сердца вплоть до взрослого состояния [3]. Кроме того, миокардин экспрессируется в субнаборе сосудистых и висцеральных типов гладкомышечных клеток. Экспрессия особенно обильна в висцеральных SMCs из желудка, мочевого пузыря и кишечника, где экспрессия миокардина предшествует экспрессии гладкомышечных генов, таких как SM22. Следует отметить однако, что myocardin не экспрессируется во всех типах развивающихся SMC и существует курьёзное расхождение между временем экспрессии myocardin и его генов мишеней в SMCs
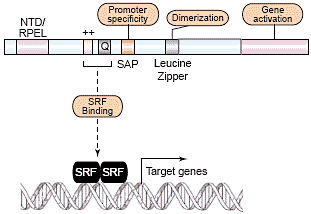 Рис. 2. Functional domains of myocardin. Myocardin contains an NTD/RPEL domain. A related domain in MAL has been shown to regulate nuclear import. A segment of the protein including the basic and
Q-rich domains mediates interaction of myocardin with SRF
homodimers. The SAP domain is required for activation of a subset
of SRF target genes, suggesting it confers promoter specificity. The
leucine zipper mediates homodimerization or heterodimerization of
myocardin with MRTFs. The transcription activation domain at the
C terminus is required for gene activation.
Рис. 2. Functional domains of myocardin. Myocardin contains an NTD/RPEL domain. A related domain in MAL has been shown to regulate nuclear import. A segment of the protein including the basic and
Q-rich domains mediates interaction of myocardin with SRF
homodimers. The SAP domain is required for activation of a subset
of SRF target genes, suggesting it confers promoter specificity. The
leucine zipper mediates homodimerization or heterodimerization of
myocardin with MRTFs. The transcription activation domain at the
C terminus is required for gene activation.
дорсальной аорты [3,37]. Эти наблюдения подтверждают существование myocardin-независимых механизмов для активации экспрессии SM генов. Являются ли MRTFs ответственными за экспрессию SM генов в отсутствие myocardin, неизвестно. Myocardin не экспрессируется в скелетно-мышечных клетках несмотря на потребность в CArG боксах для экспрессии многочисленных скелетно-мышечных структурных генов.
MRTF-A и -B экспрессируются повсеместно во время эмбриогенеза и у взрослых [4], указывая на то, что эти факторы м. вовлекаться и в др. биологические процессы помимо дифференцировки мышечных клеток.
Myocardin is necessary and sufficient for
smooth muscle cell differentiation
Экспрессия кардиальных генов м.б. блокирована у эмбрионов
Xenopus инъекциями мРНК, кодирующей доминантно негативную мутацию myocardin мыши в вентральный бластомер, предназначенный формировать сердце [3]. Сходным образом, экспрессия доминантно негативного myocardin в линии P19CL6 клеток тератокарциномы ингибирует кардиогенез [52]. Т.к. это исследование уеазывает на существенную роль myocardin в кардиогенезе, то , возможно, что доминантно негативный мутантный myocardin д. просто нарушать активность SRF и тем самым блокировать кардиогенез посредством неспецифического механизма, не связанного с функцией белка дикого типа. Согласуется с этим заключением то, что myocardin действует как регулятор экспрессии кардиальных генов у
Xenopus, a
Xenopus гомолог мышиного myocardin, как было показано с помощью
RNAi, необходим для развития сердца (E Small, D Wang, E Olson, P Kreig,
unpublished). Напротив, неправильная экспрессия myocardin у эмбрионов
Xenopus с помощью инъекций мРНК вызывает эктопическую активацию кардиальных генов. Более того, трансгенные лягушки, которые экспрессируют myocardin в нейронах спинного мозга под контролем специфичного для нейронов промотора β-tubulin, обнаруживают экспрессию кардиальных генов в спинном мозге. Myocardin существенен также для активации экспрессии кардиальных генов в анимальных шапочках
Xenopus in vitro . Хотя представленные выше эксперименты демонстрируют важность myocardin для активации экспрессии кардиальных генов, но очевидно, что активируется только субнабор кардиальных генов, это указывает на то, что дополнительные факторы м.б. необходимы для активации всего кардиального фенотипа.
В противоположность способности myocardin активировать экспрессию кардиальных генов у эмбрионов
Xenopus и анимальных шапочках, эктопическая избыточная экспрессия myocardin в культивируемых фибробластах млекопитающих не способна активировать экспрессию кардиальных генов. Вместо этого myocardin активирует SMC гены [36-39,40]. Сходным образом. форсированная экспрессия myocardin в ES клетках также достаточна для активации гладкомышечных, но не экспрессии кардиальных мышечных генов [37]. MRTF-A является столь же эффективным как и myocardin в индукции экспрессии SM генов в трансфицированных фибробластах [38], это выглядит парадоксально, учитывая, что MRTF-A экспрессируется в широком круге типов клеток, который не экспрессируют SM генов. Вообще-то активация SM генов с помощью MRTF-A нуждается в пороге экспрессии, выше эндогенного уровня или вообще активность MRTF-A д. репрессироватся в немышечных типах клеток
in vivo. Если myocardin (и MRTFs) м. активировать эндогенные SM гены в трансфицированных фибробластах или ES клетках, то неясно отражает ли это стабильное превращение таких клеток в SM фенотип или просто направленную активацию специфических SM генов мишеней для SRF.
Дальнейшие доказательства роли myocardin и MRTFs в дифференцировке SMC получены благодаря находкам, что доминантно негативный myocardin или rMRTF мутации или помехи мРНК для myocardin предупреждают дифференцировку SMCs [38,39]. Сходным образом, доминантно негативный MKL также блокирует дифференцировку линии C2C12 скелетно-мышечныхы клеток
in vitro [41], указывая тем самым, что эти факторы необходимы для SRF-зависимой активации скелетно-мышечных генов тоже.
Почему myocardin активирует кардиальные гены в некоторых условиях и SM гены в др.? Мы склоняемся к возможности, что миогенная активность myocardin испытывает влияние со стороны др. факторов и сигнальных механизмов. В сложных условиях у эмбрионов
Xenopus д. существовать дополнительные транскрипционные факторы и сигнальные события, которые являются пермиссивными для экспрессии кардиальных генов. Вообще-то эти влияния отсутствуют в контексте трансфекционных подходов культуры ткани млекопитающих. Напротив, или в дополнение, фибробласты млекопитающих м. экспрессировать негативные регуляторы кардиогенеза. В этой связи предыдущие исследования показали. что кардиальный фенотип является рецессивным и м. замалчиваться у гетерокарионов между кардиомиоцитами и фибробластами [42]. Сообщалось также, что myocardin м. активировать экспрессию кардио-специфичного гена ANF в трансфицированных клетках скелетных мышц [36]. Могут ли транскрипционные факторы, экспрессирующиеся в клетках скелетных мышц, обеспечивать частичную поддержку кардиального фенотипа, остается выяснить.
Ключевым вопрос, является ли SRF облигатным партнёром для
myocardin. Производили трансфекцию SRF нулевых ES клеток, в которых myocardin не способен активировать ни эндогенные SM гены, ни SRF-зависимые репортерные гены [4,37]. Добавление экзогенного SRF восстанавливало активность myocardin в таких клетках. Более того, только ДНК связывающий домен SRF необходим для восстановления myocardin-зависимой транскрипции в SRF нулевых ES клетках [37], что подтверждает заключение о том, что myocardin с этой областью SRF необходим и достаточен, чтобы активировать гены мишени для SRF. Конечно, эти результаты не исключают возможности, что миокардин может также ассоциировать и с др. транскрипционными факторами на SRF-независимых генах. Неожиданным оказалось то, что развитие сердца, по-видимому, оставалась неизменным у нокаутных по миокардину мышей, которые погибали на ~E11.5 из-за отсутствия сосудистых SMCs [43]. Т.к. и MRTF-A и -B также экспрессируются в эмбриональном сердце во время развития мышей, то они скорее всего компенсируют отсутствие функции myocardin. Мыши, лишенные MRTF-A жизнеспособны, тогда как делеция MRTF-B вызывает раннюю эмбриональную гибель [44].
Интересна возможная роль myocardin и MRTFs в кардиальной гипертрофии, Многочисленные стрессовые сигналы ведут к гипертрофическому росту сердца, это сопровождается активацией SRF-зависимой транскрипции. Стоит отметить в этой связи, что форсированная экспрессия SRF в сердце оказывается достаточной для индукции гипертрофической кардиопатии [45].
Более того, недавние исследования показали, что экспрессия myocardin усиливается в сердце человека после инфаркта [46], это могут объяснить изменения в экспрессии SRF-зависимых генов в таких случаях.
Signaling pathways leading to SRF
Сыворотка и очищенные ростовые факторы стимулируют активность SRF посредством двух независимых путей, одного, зависящего от фосфорилирования TCFs с помощью MAP киназного каскада, и др., зависящего от передачи сигналов Rho и динамики актина [47] (Figure 3). Доминантно негативные мутации MRTF или ингибирование экспрессии MRTF-A/B с помощью RNA interference, как было показано, специфически ингибируют SRF-зависимую активацию промотора c-fos в ответ на сыворотку и RhoA, тогда как TCF-обеспечиваемая активация оказывалась не затронутой потерей активности MRTF [30]. Ингибирование MRTF-A и -B необходимо для максимальной супрессии RhoA-зависимой активации SRF, это подтверждает, что действие этих факторов перекрывается.
Miralles et al. показали, что MAL (MRTF-A) секвестрируется в цитоплазме serum-starved фибробластов и подвергается зависимому от сыворотки ядерному импорту посредством механизма, зависящего от передачи сигналов RhoA [32]. N-терминальные RPEL мотивы необходимы для сигнал-зависимого ядерного импорта и ассоциируют косвенно с G-actin. Хотя отсутствуют доказательства прямого взаимодействия актина с RPEL мотивом MAL, но G-actin четко репрессирует ядерный импорт MAL и может быть иммунопреципитирован с MAL, следовательно, должны существовать один или несколько белков, которые связывают actin с MAL. Идентификация таких белков является важной задачей будущего. Эти находки подчеркивают, что MRTFs являются ключевой связью между передачей сигналов Rho-actin и SRF-зависимой транскрипцией. В отличие от MRTFs, которые подвергаются сигнал-зависимому ядерному импорту, myocardin локализуется постоянно
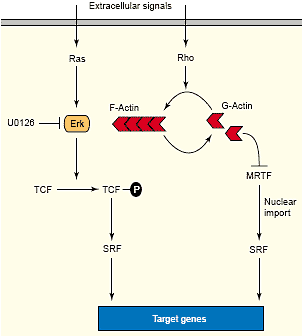 Рис. 3. Parallel signaling pathways leading to SRF activation. Growth factor signaling activates Ras/MAP kinase cascades that culminate with ERKs and phosphorylation of TCFs. Phospho-TCFs are recruited by SRF to specific SRF binding sites. Rho signaling promotes actin treadmilling. G-actin inhibits the activity of MRTF by sequestering
it in the cytoplasm. Actin polymerization releases MRTF for nuclear
import.
Рис. 3. Parallel signaling pathways leading to SRF activation. Growth factor signaling activates Ras/MAP kinase cascades that culminate with ERKs and phosphorylation of TCFs. Phospho-TCFs are recruited by SRF to specific SRF binding sites. Rho signaling promotes actin treadmilling. G-actin inhibits the activity of MRTF by sequestering
it in the cytoplasm. Actin polymerization releases MRTF for nuclear
import.
в ядре [3], это, по-видимому, отражает нечувствительность его N-терминального домена к динамике актина. MRTFs, как было показано, подвергаются сигнал-зависимым изменениям в фосфорилировании [32]. Специфические места фосфорилирования и их потенциальные вклады в SRF-зависимую транскрипцию пока не установлены.
В соответствии со стимулирующим эффектом передачи сигналов Rho на активность SRF, дифференцировка SMCs усиливается за счёт активаторов передачи сигналов Rho и полимеризации актина [48]. Недавние исследования подтвердили, что зависимость дифференцировки SM от передачи сигналов Rho отражает необходимую роль MRTFs в этом процессе [33].
Antagonists of myocardin/MRTF activity
В принципе, репрессия активности SRF должна противодействовать активности myocardin и MRTFs. Существуют многочисленные механизмы, с помощью которых такой антагонизм может осуществляться. Небольшой homeodomain-only protein HOP, как было установлено, супрессирует активность SRF путем вмешательства в способность SRF связывать ДНК. Соотв., HOP может супрессировать myocardin-зависимую активацию SRF [23,24]. Сходным образом транскрипционные факторы GATA действуют как мощные репрессоры активности SRF за счёт многочисленных механизмов [49].
Switch between differentiation and
proliferation of smooth muscle cells
Как SRF осуществляет выбор между мышце-специфическими и регулирующими рост генами не очень понятно. Любой предполагаемый механизм активации транскрипции SM генов д. объяснять способность сигналов от ростовых факторов обратимо супрессировать программу дифференцировки. В этой связи обратимая ассоциация myocardin с SRF возможно и обеспечивает механизм пластичности фенотипов SM [50].
Многие, но не все, SM гены содержат TCF связывающие сайты вблизи соседствующих CArG боксов. В SMCs, myocardin и TCFs, как было показано, ассоциируют с SRF внутри нативного хроматина взаимно исключающим и сигнал-зависимым образом [50]. Интересно, что myocardin и MRTFs содержат короткий гидрофобный участок из аминокислот с потенциалом принимать вторичную структуру, сходную с той, что в TCF B-box, это обеспечивает связывание SRF [50]. Более того, Elk-1 и myocardin соединяются с одним и тем же docking сайтом на SRF; их связывание является взаимно исключающим, так что ассоциация myocardin с SRF активирует экспрессию SM-специфических генов, тогда как формирование TCF/SRF комплекса репрессирует экспрессию SM генов и активирует экспрессию генов клеточного роста [50] (Figure 4).
Кажется парадоксальным, что Elk-1 может активировать гены, индуцибельные фактором роста, такие как c-fos, при этом он ингибирует экспрессию SM генов мишеней для myocardin. Одним из объяснений этого наблюдения может быть то, что Elk-1 (и др. TCFs) является довольно слабым транскрипционным активатором по сравнению с myocardin. Т.о., его замещение миокардина в
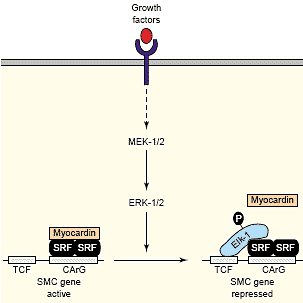 Рис. 4. A model for the opposing roles of myocardin and TCFs in the control of SMC genes. Growth factor signaling activates MEK-1/2, which activate ERK-1/2, which phosphorylates TCFs, such as Elk-1.
Phospho-Elk-1 is recruited to a subset of SMC genes where it can
displace myocardin with consequent down-regulation of transcription.
Рис. 4. A model for the opposing roles of myocardin and TCFs in the control of SMC genes. Growth factor signaling activates MEK-1/2, which activate ERK-1/2, which phosphorylates TCFs, such as Elk-1.
Phospho-Elk-1 is recruited to a subset of SMC genes where it can
displace myocardin with consequent down-regulation of transcription.
связке с SRF д. приводить к снижению экспрессии генов мишеней миокардина. Т.к. TCF сайты не содержат совсем промоторов SMC генов, то д. существовать дополнительные механизмы для негативной регуляции активности myocardin в условиях фенотипического модулирования SMC. Мутации TCF сайта в промоторе SM22 делают ген нечувствительным к репрессивным эффектам сыворотки или PDGF [50]. Более того, промотор, лишенный TCF сайта, продолжает экспрессироваться в эмбриональном сердце
in vivo в течение нескольких дней после того, как экспрессия промотора дикого типа подавляется. Это указывает на то, что конкуренция между TCFs и миокардом является важной для нормального онтогенетического контроля экспрессии SM генов в развивающемся сердце
in vivo.
Conclusions and questions for the future
The discovery of myocardin and MRTFs has provided
new models and mechanisms to account for the seemingly
paradoxical roles of SRF in the control of cell proliferation
and myogenesis, and has raised many intriguing questions
for the future. For example, how is the expression of
myocardin controlled within the earliest progenitors of
the cardiac and SM lineages? What are the functions
of MRTFs in vivo and to what extent are they redundant
with myocardin? How do myocardin and MRTFs contribute to the heterogeneity of SMCs? What are the transcriptional targets of myocardin/MRTFs in different cell types, and how do these coactivators discriminate
between genes involved in cell proliferation, myogenesis,
and other cellular functions. Do myocardin and MRTFs
associate with transcription factors in addition to SRF?
How are the activities of myocardin and MRTFs regulated
in response to extracellular signaling, and how do
MRTFs sense changes in actin dynamics? Myocardin and
MRTFs do not associate with MEF2, a MADS box
transcription factor that binds a similar site to SRF and
associates with cell type-restricted and signal-responsive
cofactors to control the expression of muscle and growth
factor-regulated genes. The similarities in mechanism of
action of SRF and MEF2 raise the possibility that cofactors
analogous to the myocardin family may also modulate
MEF2 activity. Given the central roles of SRF in the
control of cell migration, adhesion, and cytoskeletal
dynamics [51], it is likely that myocardin and MRTFs
will be intimately involved in these processes. Finally, in
light of the involvement of SRF in many pathological
states, it will be of interest to further explore the potential
contributions of myocardin and MRTFs to disease pathologies.
Update
Following submission of this review, the following
publications [53–55] provided further insight into the
functions of the myocardin family.
Сайт создан в системе
uCoz