В США болезни сердца являются убийцей номер один у взрослых. Др. 5 миллионов живут с недостаточностью сердечной функции (Thom et al., 2006). Стволовые клетки обладают чрезвычайным потенциалом в регенеративной медицине и информация о кардиогенезе от клеток предшественников во время эмбриогенеза может сформировать базу репрограммированных клеток для терапевтического использования (Srivastava, Ivey, 2006).
Полная и аккуратная картина кардиогенеза только начинает проявляться (Buckingham et al., 2005). Два самостоятельных мезодермальных поля сердца с общим происхождением возникают, чтобы внести свой dkrl в развивающееся сердце специфическим пространственно-временным способом. Хорошо изученное "первичное" или "первое" поле сердца (FHF) происходит из клеток передней части латеральной пластинки мезодермы, которая формирует форму в виде полумесяца приблизительно на ст. Е7.5 у эмбрионов мышей, что соответствует примерно 2 неделям беременности у людей (Рис. 1). На ст. Е8.0 у мышей или 3 недели у людей эти клетки сливаются вдоль вентральной срединной линии. чтобы сформировать первичную сердечную трубку, состоящую из внутреннего слоя эндокардиальных клеток и наружного слоя миокардиальных клеток, разделенных внеклеточным матриксом, через который осуществляются реципрокная передача сигналов между двумя слоями.
Предыдущие отслеживания клонов с использованием техники мечения краской, показали, что клетки вдоль передне-задней оси сердечной трубки д. вносить свой вклад в специфические камеры будущего сердца (Srivastava, Olson, 2000). Однако, эти исследования не смогли установить клонального вклада индивидуальных клеток (Meilhac et al., 2004). Более недавние исследования с использованием Cre-lox rkjyfmyjuj анализа и соотв. клонального анализа связей показали, что сердечная трубка, происходящая из FHF, может преимущественно обеспечивать каркас, на котором клетки из вторичного поля сердца (SHF) мигрируют и строят реквизиты кардиальных камер (Buckingham et al., 2005). SHF маркируется с помощью LIM-гомеодоменового транскрипционного фактора Islet1 (Isl1) и представляет собой дорсо-медиальный аспект кардиогенной пластинки, в то время как FHF представлено вентральным аспектом кардиогенной пластинки (Cai et al., 2003) (Рис. 1). FHF дифференцируется на стадии кардиального серпа, в то время как дифференцировка SHF относительно задерживается и происходит, когда второй клон мигрирует а соединение дифференцирующихся клеток первого клона. Оба клона, , по-видимому, регулируются сложной сетью позитивных и негативных сигналов с вовлечением членов bone morphogenetic (Bmp), sonic hedgehog (Shh), Fibroblast growth factor (Fgf), Wnt и Notch белков. Такие сигналы часто возникают от соседней энтодермы, хотя их точная природа и роль остаются неизвестными (Zaffar, Frasch, 2002). SHF клетки остаются в недифференцированном состоянии предшественников вплоть до инкорпорации в сердце, т.к. они находятся вблизи ингибирующих Wnt сигналов, которые исходят из срединной линии.
Как только формируется сердечная трубка, то SHF клетки мигрируют в срединную линию и располагают сами себя дорсальнее сердечной трубки в фарингеальной мезодерме (Рис. 1). После
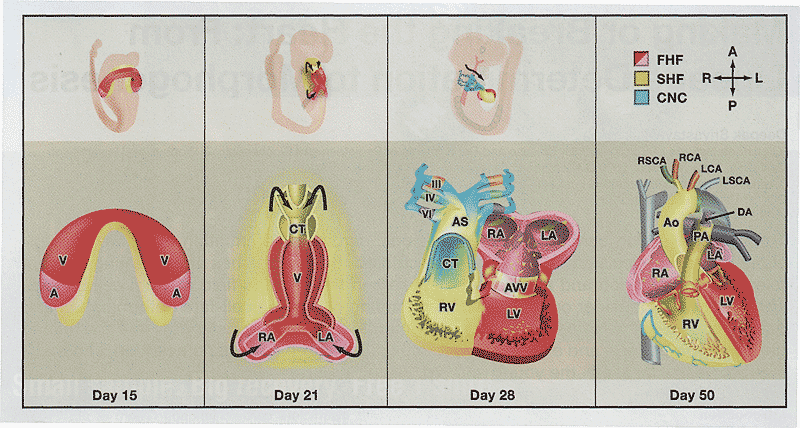 Mammalian Heart Development
Oblique views of whole embryos and frontal views of cardiac precursors during human cardiac development are shown. (First panel) First heart field (FHF) cells form a crescent shape in the anterior embryo with second heart field (SHF) cells medial and anterior to the FHF. (Second panel) SHF cells lie dorsal to the straight heart tube and begin to migrate (arrows) into the anterior and posterior ends of the tube to form the right ventricle (RV), conotruncus (CT), and part of the atria (A). (Third panel) Following rightward looping of the heart tube, cardiac neural crest (CNC) cells also migrate (arrow) into the outflow tract from the neural folds to septate the outflow tract and pattern the bilaterally symmetric aortic arch arteries (III, IV, and VI). (Fourth panel) Septation of the ventricles, atria, and atrioventricular valves (AW) results in the four-chambered heart. V, ventricle; LV, left ventricle; LA, left atrium; RA, right atrium; AS, aortic sac; Ao, aorta; PA, pulmonary artery; R§CA, right subclavian artery; LSCA, left subclavian artery; RCA, right carotid artery; LCA, left carotid artery; DA, ductus arteriosus.
Mammalian Heart Development
Oblique views of whole embryos and frontal views of cardiac precursors during human cardiac development are shown. (First panel) First heart field (FHF) cells form a crescent shape in the anterior embryo with second heart field (SHF) cells medial and anterior to the FHF. (Second panel) SHF cells lie dorsal to the straight heart tube and begin to migrate (arrows) into the anterior and posterior ends of the tube to form the right ventricle (RV), conotruncus (CT), and part of the atria (A). (Third panel) Following rightward looping of the heart tube, cardiac neural crest (CNC) cells also migrate (arrow) into the outflow tract from the neural folds to septate the outflow tract and pattern the bilaterally symmetric aortic arch arteries (III, IV, and VI). (Fourth panel) Septation of the ventricles, atria, and atrioventricular valves (AW) results in the four-chambered heart. V, ventricle; LV, left ventricle; LA, left atrium; RA, right atrium; AS, aortic sac; Ao, aorta; PA, pulmonary artery; R§CA, right subclavian artery; LSCA, left subclavian artery; RCA, right carotid artery; LCA, left carotid artery; DA, ductus arteriosus.
образования петли в правую сторону сердечной трубки клетки SHF пересекают фарингеальную мезодерму в передней и задних порциях, занимая большую порцию тракта оттока, будущего правого желудочка и предсердий (Сai et al., 2003). Предшественники левого желудочка рыхло пронизываются клетками SHF и , по-видимому, в основном происходят из FHF. Затем внутри сердца FHF и SHF клетки , по-видимому, пролиферируют в ответ на сигналы. происходящие из эндокарда, такие как neuregulin, и эпикардиальные сигналы, зависимые от ретиноевой кислоты, хотя эти механизмы остаются плохо изученными (Olson, 2004). Количество кардиальных миоцитов во время развития также может регулироваться homeodomen-only protein (Hop), который действует иерархически ниже Nkx2-5 (Shin et al., 2002; Chen et al., 2002).
Transcriptional Regulation of Cardiac Precursors
FHF и SHF регуляция связана с использованием многочисленных сигнальных и транскрипционных каскадов (Рис. 2). Факторы, секретируемые передней частью сердечной трубки, могут служить как хемоаттрактантые сигналы, которые индуцируют миграцию клеток SHF, хотя природа таких молекул остается неизвестной. Экспрессия Isl1 подавляется, когда клетки предшественники экспрессируют маркеры кардиальной дифференцировки, но Isl1 необходим для клеток, происходящих из SHF, чтобы внедриться в сердце (Cai et al., 2003). Как Isl1 регулирует SHF клетки предшественники ещё предстоит выяснить. Интересно, что Isl1-позитивные клетки маркируют ниши недифференцированных клеток кардиальных предшественников в постнатальном сердце (Laugwitz et al., 2005), подтверждая, что понимание регуляции пула происходящих из SHF клеток предшественников может быть успешным для разработки подходов для репарации сердца.
Открытие SHF привело к реинпретации находок у мышей, лишенных критических регуляторных белков и у трансгенных мышей подавляющих энхансеры генов, экспрессируемых в сердце. Т.к. молекулярные аспекты кардиогенеза были впервые выявлены декаду тому назад, специфические для правого желудочка энхансеры были найдены для некоторых генов. широко экспрессирующихся в развивающемся сердце (Firulli, Olsen, 1997). Затем было установлено, что правый желудочек богат транскрипционным фактором Hand2 (известным также как dHAND), необходимым для экспансии правого желудочка. Учитывая, что Hand2 экспрессируется на высоком уровне в SHF, то гипоплазия правого желудочка наблюдается при нарушении Hand2 у мышей и скорее всего связана с неспособностью клеток SHF расширяться в правый желудочек. Сходным образом, гипоплазия правого желудочка у мышей, лишенных Mef2c, мишени для Isl1, Gata4, Foxh1 и Tbx20 в SHF (Dodou et al., 2004; von Both et al., 2004), также может быть связана с дефектами развития SHF. В самом деле, многие центральные транскрипционные регуляторы кардиального развития в FHF, включая Nkx2-2 и Gata4, обнаружены в SHF и могут целенаправленно нарушать развитие SHF (Zeisberg et al., 2005) (Рис. 2). Пути, регулирующие дифференцировку клеток SHF, могут составлять базу для индукции кардиальных клеток из клеток предшественников.
Значение транскрипционной регуляции SHF подчеркивается кардиальными дефектами при синдроме DiGeorge. При этом синдроме, который обычно связан с делецией 22q11, транскрипционный фактор ЕИЧ1 вызывает кардиальные черепно-лицевые нарушения. TBX1, центральный регулятор транскрипции SHF, необходим для собственно развития миокарда кардиального тракта оттока (Hu et al., 2004; Xu et al., 2004). Shh необходим для поддержания экспрессии Tbx1 в SHF посредством forkhead-содержащих транскрипционных факторов, которые непосредственно регулируют Tbx1 (Garg et al., 2001; Yamagishi et al., 20030. Соотв. мыши, лишенные Shh, Foxc1 и Foxc2 и Tbx1 имеют сходные дефекты в тракте оттока сердца (Kume et al., 2001; Yamagishi et al., 2003). Tbx1 регулирует миокрад тракта оттока и продукцию ростовых факторов, таких как Fgf8, который секретируется и активирует рецепторы на соседних клетках. производных нервного гребня, чтобы повлиять на их дифференцировку (Abu-Issa et al., 2002; Frank et al., 2002; Hu et al., 2004).
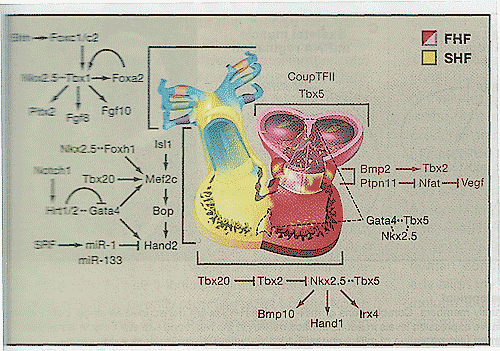 Figure 2. Pathways Regulating Region-Specific Cardiac Morphogenesis
A partial list of transcription factors, signaling proteins, and miRNAs that can be placed in pathways that influence the formation of regions of the heart is shown. Positive influences are indicated by arrowheads, and negative effects by bars. Physical interactions are indicated by dashed lines between factors. In some cases relationships of proteins are unknown. Pathways regulating neural crest cells have been reviewed elsewhere (Stoller and Epstein, 2005). FHF, first heart field; SHF, second heart field.
Figure 2. Pathways Regulating Region-Specific Cardiac Morphogenesis
A partial list of transcription factors, signaling proteins, and miRNAs that can be placed in pathways that influence the formation of regions of the heart is shown. Positive influences are indicated by arrowheads, and negative effects by bars. Physical interactions are indicated by dashed lines between factors. In some cases relationships of proteins are unknown. Pathways regulating neural crest cells have been reviewed elsewhere (Stoller and Epstein, 2005). FHF, first heart field; SHF, second heart field.
Hand2 и родственный Hand1 экспрессируются в SHF; однако, они экспрессируются также в FHF, при этом Hand1 обнаруживается на более высоком уровне в левом желудочке. Экспрессия Hand1 зависит от Nkx2-5 в левом желудочке, указывая тем самым, что Nkx2-5 является критическим также и для FHF (Biben, Harvey, 1997). Хотя нарушение tinman, ортолога Nkx2-5 у мух, приводит к полной потере кардиальных клеток (Bodmer, 1993), делеция Nkx2-5 у мышей менее тяжелая с летальностью на ст. Е9.5 после инициального образования сердечной трубки (Lyons et al., 1995; Tanaka et al., 1999). Однако, потеря Nkx2-5 и Hand2 вызывает полную неспособность экспансии желудочков у мышей (Yamagishi et al., 2001). У рыб данио рерио и плодовых мух отсутствие одиночного Hand ортолога делает невозможным расширение пула сравнимых вентрикулярных предшественников, это согласуется с данными, полученными на мышах (Yelon et al., 2000; Han et al., 2006).
Сохранение атриальных предшественников у мутантов Hand мышей и рыб подтверждает, что отдельные популяции предшественников могут вносить вклад в предсердия. В самом деле, мыши с отсутствием ядерного рецептора CoupTFII лишены атриальных миоцитов (Pereira et al., 1999)> также как и у мышей, лишенных Tbx5 (Bruneau et al., 2001). Разные аспекты атриальной или вентрикулярной экспрессии генов, , по-видимому, регули he.ncz с помощью Irx4 из семейства Iroquois транскрипционных факторов (Bao et al., 1999).
Эпигенетические факторы могут также вносить вклад в дифференцировку кардиомиоцитов и морфогенез камер. Нарушения хроматин ремоделирующего белка Smyd1 (или Bop) дают фенотип, напоминающий мутантов Hand2: маленький сегмент правого желудочка и слабое развитие миокарда левого желудочка (Gottlieb et al., 2002). Smyd1 содержит SET домен, который обладает метилтрансферазной активностью и рекрутирует также активность гистоновой деацетилазы (HDAC), которые вместе репрессируют гены. Активность Smyd1 необходима для экспрессии Hand2 в кардиальных предшественниках посредством неизвестных посредников. Интересно, что Smyd1 является непосредственной мишенью для Mef2c (Phan et al., 2005), указывая тем самым, что Isl1, Mef2c и Hand скоординированно регулируют развитие вентрикулярных кардиомиоцитов (Рис. 2). Прямая роль Hands была также продемонстрирована с помощью неспособности к вентриркулярному росту у мутантных мышей, лишенных HDAC5 и HDAC9 (Chang et al., 2004b). Этот и др. пути, , по-видимому, регулируются с помощью мышечно-специфических членов комплекса SWI/SNF? Baf60c (Licjert et al., 2004), подтверждая, что транскрипционная активность кардиальную ДНК связывающих белков тонко регулируется посредством эпигенетических событий.
MicroRNA Regulation of Cardiomyocyte Differentiation
Хотя транскрипционные и эпигенетические события регулируют многие критические кардиальные гены, трансляционный контроль с помощью малых не кодирующих РНК, таких как miRNAs, оказался др. механизмом тонкой настройки дозы ключевых белков во время кардиогенеза (Zhao et al., 2005; Kwon et al., 2005). miRNAs геномно кодируемые в 20-22 нуклеотида РНК, которые нацелены на мРНК для ингибирования трансляции или деградации с помощью многих из тех же самых путей, что используются smal interfering (si)RNAs (He, Hannon, 2004; Ambros, 2004). Функция и РНК мишени известны для немногих для более 400 идентифицированных у человека miRNAs.
miR-1-1 и mir-1-2 высоко законсервированы и специфически экспрессируются в развивающихся клетках предшественниках сердечной и скелетных мышц (Zhao et al., 2005). miR-1-1 первоначально встречается в большом количестве в атриальных предшественниках прежде чем стать повсеместными в сердце; miR-1-2 специфична для желудочков, что подтверждается её камер-специфическими эффектами. Интересно, что их экспрессия контролируется с помощью хорошо изученной транскрипционной регуляторной сети, которая способствует мышечной дифференцировке (Рис. 3). Кардиальная экспрессия зависит от serum response factor (SRF), а скелетно-мышечная экспрессия нуждается в миогенном транскрипционном факторе MyoD и Mef2. SRF рекрутирует мощный коактиватор, myocardin, на специфические для сердца и гладких мышц гены, которые контролируют дифференцировку (Wang et al., 2001). Согласуется с ролью в дифференцировке избыточная экспрессия miR-1 в развивающемся сердце мыши снижает экспансию вентрикулярных миоцитов при этом лишь немногие пролиферирующие кардиомиоциты остаются в клеточном цикле. miR-1 и miR-133a транскрибируются в виде полицистронной мРНК и коэкспрессируются в сердце и скелетных мышцах, обнаруживая общую транскрипционную регуляцию (Рис. 3). Как и в кардиальных мышцах, miR-1 способствует дифференцировке скелетных миобластов в культуре, но, что интересно, miR-133a ингибирует дифференцировку и способствует пролиферации миобластов (Chen et al., 2006).
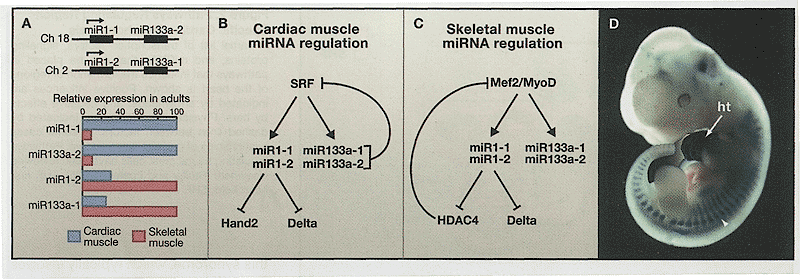
Полное понимание того, как miRNAs регулируют биологические события, ограничивается неспособностью идентифицировать мРНК мишени. Эта неспособность отражает ограниченную степень тождественности последовательностей, необходимую для распознавания мишеней, обычно нуклеотидов 2-7 на 5' конце miRNA. In silico поиск, базирующийся на тождественности последовательностей часто приводит к обнаружению нескольких сотен потенциальных мишеней, что делает трудной оценку. Мишени для miRNA идентифицированные благодаря генетическому скринингу у мух и червей постоянно располагаются в доступных доменах, определяемых по свойствам вторичной структуры РНК и свободной энергии связывания {Zao et al., 2005). Включение этих свойств в биоинформационные подходы существенно улучшает специфичность предсказаний мишеней и было использовано для идентификации Hand2 мРНК в качестве мишени для miR-1. Этот результат подтверждает, что точная регуляция уровней Hand2 белка может быть использована для контроля баланса между пролиферацией и дифференцировкой кардиомиоцитов (Рис. 3). HDAC4, ингибитор Mef2, также может быть мишенью miR-1 в скелетных мышцах, способствуя эффектам Mef2 во время дифференцировки (Chen et al., 2006).
Единственный ортолог miR-1 у мух участвует рано в запуске клеточной дифференцировки кардиальных предшественников и позднее в поддержании экспрессии кардиальных генов et al., 2005). Легкий miR-1 фенотип также указывает на изменчивость фенотипа (Sokol, Ambros, 2005), хотя нокдаун активности miR-1 с помощью )-methyl-модифицированных антисмысловых олигонуклеотидов приводит всегда к летальности (leaman et al., 2005). miR-1 у vs[ регулирует Notch лиганд Delta (Kwon et al., 2005), потенциально объясняет участие miR-1 в происхождении дифференцированных кардиальных клеток из эквивалентной группы клеток предшественников (Рис. 3). Т.о., miR-1 может быть мышце-специфической miRNA, которая управляет клетками предшественниками в направлении приобретения судьбы кардиальных клеток путем регуляции медиаторов дифференцировки.
Complex Regulation of Cardiac Morphogenesis
Хотя вклад клеточных клонов в развитие сердца достаточно хорошо изучен, последующие события, такие как интеграция множественных клеточных типов, формирование камер и формирование паттерна определенных областей сердца. еще предстоит выяснить. Некоторые аспекты описаны ниже, др. рассмотрены в др. местах (Olson, 2004; Parmaceck, Epstein, 2005). Отметим, что развитие проводящей системы сердца здесь не обсуждается, хотя это существенно для нормальной функции сердца (rev. Mikawa et al., 2003).Др. находки проливают свет на важность интеграции между специализированными проводящими клетками и миокардом (Costantini et al., 2005).
Dorsal-Ventral Polarity
Различимая дорсо-вентральная (DV) полярность возникает в первичной сердечной трубке. Т.к. сердечная трубка образует петлю вправо, то вентральная сторона трубки ротирует, чтобы оказаться на наружном изгибе петлеобразного сердца, а дорсальная поверхность формирует внутренний изгиб. Наружный изгиб становится местом активного роста, тогда как ремоделирование внутреннего изгиба контролирует расположение трактов притока и оттока сердца. Модель, согласно которой индивидуальные камеры выпячиваются ("balloon") от наружной курватуры сегментным способом, предложена Christoffels et al., (2004). Согласно этой модели многочисленные гены, включая Hand1 и саркомерный белок Serca2, экспрессируются специфически на наружном изгибе сердца (Ишиутб Harvey, 1997; Thomas et al., 1998). Также благодаря сложной транскрипционной сети, уникальные качественные особенности клеток внутреннего изгиба детерминируются с помощью Tbx2-обусловленной репрессии генов, обычно обнаруживаемых на наружном изгибе (Harrelson et al., 2004). Др. Tbox транскрипционный фактор, Tbx20, репрессирует активность Tbx2 на наружной курватуре, когда она расширяется в кардиальные камеры, чтобы установить формирование регионального паттерна расширяющегося или ремоделируемого миокарда (Stennard et al., 2005; Takeuchi et al., 2005; Cai et al., 2005; Singh et al., 2005). Ремоделирование внутренней курватуры делает возможной миграцию тракта притока направо, а тракта оттока налево., облегчая тем самым собственно расположение и разделение лево- и правостороннего кровообращения. Помимо своей роли в репрессии Tbx2, Tbx20 затрагивает экспансию FHF- и SHF-производных клеток и необходим для развития тракта оттока, возможно за счет регуляции Nkx2-5 и Mef2c (Takeuchi et al., 2005).
Left-Right Asymmetry
Дефекты ремоделирования внутренней изогнутости могут лежать в основе ВПС у людей, из-за несоответствующего расположения предсердий, желудочков и тракта оттока. Сюда входят и ситуации, когда оба атрио-вентрикулярных клапана сообщаются с левым желудочком (double-inlet left ventricle) или когда аорта и пульмональная артерия обе выходят из правого желудочка (double-outlet right ventricle), как это наблюдается у многих мутантных модельных мышей (rev. Franco, Campione, 2003). Такие дефекты часто наблюдаются совместно с аномалиями в детерминации лево-правосторонней асимметрии. Т.о., лево-правосторонние решения могут влиять на направление изгиба сердечной петли и собственно на расположение камер, скорее всего за счет регуляции генной экспрессии вдоль внутренней или наружной курватуры и вентральной в отлчие от дорсальной поверхности развивающегося сердца.
Учитывая, что элегантная молекулярная сеть регулирует лево-правостороннюю асимметрию плана тела (Palmer, 2004) необходимо отметить лишь взаимоотношения между лево-правосторонней асимметрией и собственно расположением кардальных камер. Каскад лево-правосторонних сигналов, включая Shh и Nodal, конвергирует на транскрипционном факторе Pitx2. Pitx2 инициально экспрессируется асимметрично вдоль лево-правосторонней оси в линейной сердечной трубке, но эта асимметрия транслируется в дорсо-вентральную полярность в петлеобразной сердечной трубке. Т.к. Pitx2 регулирует клеточную пролиферацию посредством cyclin D2 и также контролирует миграцию клеток (Kioussi et al., 2002), то он может связывать сигналы. регулирующие направление и процесс кардиального петлеобразования. Внутри определенных субдоменов регуляция Pitx2 с помощью Tbx1 интегрирует транскрипционные пути, контролирующие морфогенез и лево-правостороннюю асимметрию (Nowotschin et al., 2006).
Regulation of the Cardiac Outflow Tract
ВПС, затрагивающие тракт оттока, аортальные дуги, ductus arteriosus и проксимальные части пульмональной артерии, составляют 20-30% от всех врожденных пороков сердца. Эта область сердца подвергается экстенсивным и сложным морфогенетическим изменениям со вкладом клеток нервного гребня и SHF. Мезенхимные клетки, происходящие из гребня нервных складок, являются существенными для собственно разделения и ремоделирования тракта оттока и аортальных дуг (rev. Hutson, Kirby, 20030. такие клетки, производные нервного гребня, мигрируют из нервных складок и сохраняют потенциал дифференцироваться во множественные типы клеток. Миграторный путь и окончательная судьба этих клеток зависит от их относительного происхождения вдоль передне-задней оси и частично регулируются с помощью Нох генов (Le Douarin et al., 2004). Клетки нервного гребня от отического примордия в третий сомит мигрируют через развивающиеся фарингеальные дуги в мезенхиму артерий аортальных дуг и в мезенхиму, которая формирует перегородку тракта оттока (Рис. 1). Этот сегмент нервного гребня часто называется кардиальным нервным гребнем.
Мутации во многих сигнальных каскадах затрагивают миграцию нервного гребня или развитие мышей, включая пути endothelin и semaphorin и вызывают дефекты тракта оттока, сходные с теми, что наблюдаются у людей (rev. Stoller, Epstein, 2005). Апоптоз некоторых клеток, происходящих из нервного гребня, , по-видимому, существенен для собственно морфогенеза, в то время как др. дифференцируются в сосудистые гладкомышечные клетки различных регионов аортальных дуг. Напр., мутации neural crest-enriched transcription factor (TFAP2β) приводят к аномальному сохранению протока arteriosus, специального сосуда аортальных дуг, важного для кардиальной физиологии плода, возможно отражающего аномальную дифференцировку гладких мышц (Satoda et al., 2000) (Рис. 1). Др. генетические мутации могут затрагивать специфические области аортальной дуги.
Нарушение развития SHF за счет мутантных генов, таких как Tbx1 и Fgf8, приводят к дефектам, сходных с теми, что возникают при нарушениях нервного гребня (Рис. 2), включая персистенцию truncus arteriosus (неспособность разделения тракта оттока сердца), неправильное расположение тракта оттока по отношению к вентрикулярным камерам и к дефектам межжелудочковой перегородки (rev. Baldini, 2005). Реципрокные взаимодействия между SHF и клетками, производными нервного гребня в тракте оттока скорее всего существенны, т.к. происходящие из SHF миокардиальные клетки соседствуют с клетками, производными нервного гребня и секретируют Tbx1-зависимым способом факторы роста, такие как Fgf8, которые влияют на клетки нервного гребня (Hu et al., 2004). Было бы интересно определить, вызываются ли дефекты тракта оттока альтерациями в миграции, дифференцировке или пролиферации SHF.
Cardiac Valve Formation
Соотв. расположение и функционирование кардиальных клапанов является существенным для разделения камер и однонаправленного тока крови через сердце. Молекулярная сеть, вовлекающая Bmp2 и Tbx2, определяет позицию клапанов относительно камер (Harrelson et al., 2004; Ma et al., 2005). Dj время раннего формирования сердечной трубки, "подушки" внеклеточного матрикса между эндокардом и миокардом вносят вклад в формирование клапанов, которое происходит на каждом из концов сердечной трубки. Реципрокная передача сигналов, обеспечиваемая частично с помощью TGF-β членов между эндокардом и миокардом в области подушек индуцирует трансформацию эндокардиальных клеток в мезенхимные клетки, которые мигрируют во внеклеточный матрикс подушек (rev. Armstrong, Bischoff, 2004). Эти мезенхимные клетки дифференцируются в фиброзную ткань клапанов и участвуют в разделении общего атриовентрикулярного канала на право- и лево-стороннее отверстие.
Smad белки являются внутриклеточными транскрипционными медиаторами передачи сигналов, инициируемой TGF-β лигандами. Smad6 специфически экспрессируется в атриовентрикулярных подушках и тракте оттока во время кардиогенеза и является негативным регулятором передачи сигналов TGF-β. Целенаправленные нарушения Smad6 у мышей приводят к утолщению и желатинизации атриовентрикулярных и полулунных клапанов, сравнимые с теми, что наблюдаются у людей с болезнью аортальных и пульмональных клапанов (Galvin et al., 2000). Сходным образом отсутствие Ptpn11 ? который кодирует протеин тирозин фосфатазу Shp-2, приводит к дисплазии клапанов оттока посредством пути, использующем рецептор эпидермального фактора роста (Срут уе al., 2000). Важность PTPN11 для врожденных болезней сердца продемонстрирована с открытием точечных мутаций в PTPN11, которые приводят к к активации Shp-2 фосфатазы у пациентов с синдромом Noonan, который обычно сопровождается стенозом пульмональных клапанов (Tartaglia et al., 2001).
Нарушения сигнальных путей, использующих транскрипционный фактор Nfatc, выявляют потребность в этом активируемом кальцием регуляторе. Nfatc экспрессируется специфически в мезенхимных предшественниках клапанов, а мыши без Nfatc неспособны к образованию кардиальных клапанов (ву la Pompa et al., 1998; Ranger et al., 1998). Передача сигналов посредством фосфатазы, calcineurin, приводит к транслокации в ядро Nfatc и сходна с вовлечением в образование кардиальных клапанов частично за счет регуляции экспрессии vascular endithelial growth factor (Vegf) в эндокарде (Chang et al., 2004a). Интересно, что мутации Ptpn11? которые активируют фосфатазу Shp-2, увеличивают осцилляции кальция и нарушают ядерную локализацию Nfatc, предоставляя возможный механизм для дефектов клапанов, наблюдаемый при синдроме Noonan (Uhlen et a., 2006).
Путь передачи сигналов Notch также участвует в развитии кардиальных клапанов. У рыб и лягушек Notch необходим для развития эндокардиальных подушек, которые вносят вклад в ткань клапанов (Timmerman et al., 2004). У людей, гетерозиготных по мутациям NOTCH1, нарушается нормальное развитие аортальных клапанов и иногда митрального клапана (Garg et al.,2005) (Tabl.1). Тяжесть болезней клапанов, ассоциируемых c мутациями
NOTCH1, широко варьирует у людей от легкой, при которой аортальные клапаны имеют два скорее, чем три створки (bicuspid аортальные клапаны) до тяжелых дефектов в открытии клапанов
in utero , приводя к нарушениям роста левого желудочка. Около 15% "нормальных" родственников детей с гипоплазией левого желудочка (синдром гипопластического лев. желудочка) имеют субклинические bicuspid аортальные клапаны (Cripe et al., 2004), указывая тем самым, что нарушение сигнального каскада NOTCH может лежать в основе спектра болезней аортальных клапанов. Мутации в JAGGED1, лиганде NOTCH, также вызывают дефекты тракта оттока, ассоциированные с аутосомно-деминантным заболеванием, синдромом Alagille (rev.Krantz et al., 1999). Семейство hairy-родственных транскрипционных репрессоров (Hrt1, Hrt2 и Hrt3) может опосредовать Notch сигнал во время развития клапанов и миокарда; однако
их мишени неизвестны (Nakagawa et al., 1999; rev. Kokubo et al., 2005).
Genetics of Human Septal Defects
Недавние находки в отношении кардиальных транскрипционных факторов NKX2-5, TBX5 и GATA4 представляют пример синергии между генетикой человека и исследованиями на модельных организмах для понимания этиологии ВПС (Табл.1). Многочисленные точковые мутации в NKX2-5 происходят в семьях с дефектами межпредсердной перегородки и прогрессирующими аномалиями кардиальной электрической проводимости (Schott et al., 1998). Ретроспективный анализ мышей, гетерозиготных по Nkx2-5 нарушениям выявляет сходный фенотип и прогрессирующую апоптическую потерю проводящих клеток, указывая тем самым на возможный механизм подобного фенотипа у людей (Biben et al., 2000; Jay et al., 2004).
Люди с синдромом Холта-Орама, результат мутаций в TBX5, имеют кардиальные аномалии, сходные с теми, что возникают при мутациях NKX2-5 (дефекты межпредсердной и межжелудочковой перегородки), а также аномалии конечностей (Mori, Bruneau, 2004). Интересно, что мутации. вызывающие дефекты в сердце и конечностях образуют кластеры в разных регионах белка, указывая тем самым, что использует разные нижестоящие гены или кофакторы, в зависимости от уникальных структурных мотивов в белке. Один потенциальный кофактор, NKX2-5, может кооперироваться, чтобы активировать общие гены мишени (Hiroi et al., 2001).
Мутации в белке GATA4, содержащем цинковые пальчики, вызывают сходные дефекты атриальной и вентрикулярной перегородок в автосомно-доминантных несиндромальных родословных у людей (Garg et al., 2003). GATA4 или родственные белки являются существенными для кардиогенеза у мух, рыб и мышей (Epstein, Parmacek, 2005). Подобно NKX2-5, GATA4 и TBX5 также формируют комплекс для регуляции нижестоящих генов, таких как тяжелая цепь миозина. В соотв. с ролью в комбинаторных взаимодействиях, семейные GATA4 точковые мутации нарушают способность GATA4 взаимодействовать с TBX5 (Garg et al., 2003). Напротив. некоторые TBX5 мутации у людей нарушают взаимодействия GATA4-ЕИЧ5б указывая тем самым на то. что они кооперируются при образовании кардиальных перегородок. GATA4, TBX5 и NKX2-5 могут формировать комплекс. необходимый для собственно рьразования кардиальных перегородок (Рис. 2). Нарушения любого из этих белков или их взаимодействий может приводить к дефектам атриальной или вентрикулярной перегородки. Гены, регулируемые этими факторами в перегородке, неизвестны, но интересно, что мутации в тяжелой цепи α-миозина человека (MYH6) являются непосредственными мишенями для GATA4, TBX5 и NKX2-5 и также вызывают дефекты межпредсердной перегородки (Ching et al., 2005).
Adult Consequence of Cardiac Malformations
Когда люди, пережившие кардиальные пороки вступают в третью и четвертую декаду своей жизни, то возникают новые кардиальные заболевания, включая аномальное электрическое проведение и снижение контрактильной функции сердца. Ранее эти "вторичные" дефекты приписывали аномальному кровотоку, обусловленному структурными аномалиями, но появились теперь доказательства. что те же самые гены, которые вызывают ВПС могут непосредственно участвовать в дисфункции сердца и нарушениях клеточного клонирования и у взрослых (rev. Srivastava, 2004).
Напр., мутации у мышей и людей в NKX2-5 вызывают онтогенетические дефекты предсердной перегородки и также нарушают электрическое проведение через сердечные камеры и могут вызывать внезапную смерть в поздней жизни (Schott et al., 1998; Pashmforoush et al., 2004). Атриовентрикулярный узел, являющийся важным местом электрических коммуникаций между предсердиями и желудочками, оказывается меньше, чем в норме у взрослых мутантных Nkx2-5 мышей. Со временем специализированные происходящие из мышц проводящие клетки теряются и замещаются фиброзной тканью, приводя к прогрессирующим дефектам электрического проведения (Jay et al., 2004).
Lh/ пример связан с аортальными клапанами. По всему миру 1-2% людей рождаются с bicuspid аортальными клапанами, обычно не проявляющими себя в детстве (Hoffman,Kaplan, 2002). Однако, треть bicuspid аортальных клапанов обнаруживают преждевременную возраст-зависимую кальцификацию, давая плохо двигающиеся, нефункциональные клапаны в позднем возрасте (Rajamannan et al., 2003). Как результат, кальцификация аортальных клапанов, является третьей причиной заболеваний сердца у взрослых и связана с 50000 замещений клапанов в год в США. Недавнее открытие, что мутации NOTCH1 у людей вызывают bicuspid аортальные клапаны и позднее их кальцификацию указывает на то, что ранняя онтогенетическая и поздняя дегенеративная болезнь могут иметь общую генетическую причину (Garg et al., 2005).
Кальцифицированные клапаны человека характеризуются эктопической экспрессией остеобласт-специфических генов, указывая тем самым на изменения клеточной судьбы мезенхимных или воспалительных клеток (Rajamannan et al., 2003). Notch1 может репрессировать центральный транскрипционный регулятор остеобластной судьбы клеток, Runx2/Cbfa1 (Ducy et al., 1997), указывая на потенциальный механизм для NOTCH1 по супрессии кальцификации ткани клапанов (Garg et al., 2005). Было бы интересно определить, существуют ли полиморфизмы
NOTCH1, ассоциированые с изменением риска кальцификации аортальных клапанов и даже сосудов, учитывая сходную экспрессию osteoblast гена в атеросклеротических сосудистых гладких мышцах (Stetz et al., 2001). Если это так, то возможны эффективные превентивные вмешательства, напр.. в виде использования статинов, понижающих уровни холестерола. известного фактора риска кальцификации.
Cardiac Stem Cell and Regenerative Approaches
Мнение, что гены, участвующие в раннем кардиогенезе могут быть повторно использованы, чтобы помочь защитить, репарировать или регенерировать сердечную мышцу понятно, оно заставляет исследовать ранние онтогенетические пути (Parmacek, Epstein, 2005).
Сообщения, что ниши из популяций малых не кардиомиоцитарных клеток в постнатальном сердце могут дифференцироваться в кардиальные мышцы и эндотелиальные клетки, подобно др. органам, могут иметь резидентный пул клеток предшественников. Существуют некоторые противоречия относительно маркеров таких клеток с частично не перекрывающимися сообщениями о Sca-1бс-kit или Abcg2-позитивных предшественниках, которые могут дифференцироваться в кардиомиоциты (rev. Leri et al., 2005). Четвертая популяция предшественников экспрессирует Isl1, это подтверждает важность соединения между постнатальными клетками предшественниками и ранними путями развития, регулирующими предшественников кардиомиоцитов (Laugwitz et al., 2005). Как было описано ранее, Isl1 экспрессируется в SHF клетках, прежде чем они дифференцируются с миоциты и подавляется после экспрессии саркомерных белков. У мышей и людей ниши Isl1-позитивных клеток в раннем постнатальном сердце могут быть остатками онтогенетических клеток предшественников среди терминально дифференцированных миоцитов (Laugwitz et al., 2005). Было бы интересно установить, могут ли эти клетки дифференцироваться в кардиомиоциты, эндотелиальные клетки и клетки проводящей системы.
Глубокое понимание сетей, управляющих пролиферацией и дифференцировкой SHF, может позволить экспансию постнатальных кардиальных предшественников для терапевтических целей. Интересно, что Shh, регулятор плюрипотентности в некоторых наборах (Kusano et al., 2005), является ранним регулятором SHF и контролирует экспрессию Tbx1 в кардиальных предшественниках (Yamagishhi et al., 2003). Может ли Shh регулировать постнатальные предшественники, неизвестно.
Кроме того, производные костного мозга и циркулирующие стволовые клетки, вводимые мышам и людям после острого инфаркта миокарда могут вызывать некоторое улучшение функции сердца; однако, это остается дискуссионным (Rev. Leri et al., 2005; Srivastava, Levy, 2006). Хотя вряд ли происходящие из костного мозга стволовые клетки дифференцируются в миоциты, они могут выполнять не клеточно автономную функцию за счет секреции паракринных факторов, которые способствуют выживаемости или неоангиогенезу, чтобы защитить и восстановить ишемический миокард (Mangi et al., 2003). Репаративная способность происходящих из костного мозга стволовых клеток у грызунов может быть повышена путем избыточной экспрессии ангиогенной киназы Akt (protein kinase B) в клетках перед введением, приводит к повышению секретируемых факторов с потенциальной выгодой для миокарда под угрозой (Gnecchi et al., 2006).
Идентификация критических секретируемых факторов может помочь избавиться от нужды в клеточной терапии в пользу специфических жизнеспособности и ангиогенных факторов во время периода острой ишемии (Gnecchi et al., 2006). Недавние исследования онтогенетического белка указывает на механизм по достижению этой цели. Thymosin β4, в 43 аминокислоты секретируемый пептид, содержит актин связывающий домен, который обильно экспрессируется в развивающемся сердце, где он может регулировать события клеточной миграции во время морфогенеза (Bock-Marquette et al., 2004). Thymosin β4 действует в комплексе с интегрин-сцепленной киназой, чтобы активировать нижестоящие события, включая фосфорилирование Akt. Системные или локальные воздействия на мышей thymosin β4 сразу же после закупорки коронарных сосудов существенно защищает гипоксичный миокард (Bock-Marquette et al., 2004), скорее всего способствуя выживанию и неоангиогенезу (Grant et al., 1999). Интересно, что thymodin обильно секретируется стволовыми клетками костнрого мозга и его секреция увеличивает многократно избыточную экспрессию Akt в стволовых клетках. происходящих из костного мозга, это указывает на существование позитивной петли обратной связи, поддерживающей продукцию thymosin β4 (Gnecchi et al., 2006). Роль тимозина и др. секретируемых белков в передаче какого-либо потенциального эффекта стволовых клеток из костного мозга неизвестна, но они обладают важным терапевтическим потенциалом.
Сайт создан в системе
uCoz