Структурная целостность DRV определяется а) его макромолекулярной композицией, б) сложными взаимодействиями между резидентными белками и в) способностью этих молекул DRV собираться в высоко организованные, трехмерные архитектуры. В большинстве тканей первичный каркас составляют коллагеновые фибриллы или сети, которые включают разнообразные Pgs и гликопротеины. Вместе они формируют специфические супрамолекулярные ансамбли для выполнения различных механических потребностей из соединительной ткани и BMs.
Proteins Involved in the Stability of Connective Tissues
Collagen fibrils. Все коллагены состоят из трех полипептидных цепочек и обладают характерными трех-спиральными collagenous (COL) и noncollagenous (NC) доменами, Кстати, 27 типов коллагенов описано у позвоночных; они могут быть сгруппированы в 9 семейств на базе их супрамолекулярных ансамблей и др. признаков (Myllyharju & Kivirikko 2004). Среди них формирующие фибриллы коллагены собираются в длинные, высоко упорядоченные полимеры. которые противостоят высоким силам растяжения. Фибриллярные коллагены синтезируются как протоколлагеновые молекулы, состоящие из длинного COL домена, фланкированного глобулярными пропептидами на N и C концах. После секреции пропептиды обычно отщепляются и коллагеновые мономеры формируют короткие фибриллярные промежуточные образования, которые постепенно вырастают в зрелые фибриллы посредством событий линейных и боковых слияний. Хотя фибриллогенез управляется прежде всего с помощью COL домена, на фибриллярный рост и межфибриллярные пространства влияют различные молекулы DRV? которые и предопределяют финальную архитектуру коллагеновых фибрилл в разных тканях (Canty & Kadler 2005). Семейство фибриллярных коллагенов включает богатые членами типы - I, II и III - и минорные в количественном отношении типы - V и XI. Коллагены типов I, III и V имеют широкое распространение в теле, тогда как коллагены II и XI в основном ограничены хрящами, стекловидным телом и текториальной мембраной внутреннего уха. Структурные и нулевые мутации этих коллагенов вызывают нарушения у людей, такие как osteogenesis imperfecta, familial arterial aneurism, определенные субтипы синдрома Ehlers-Danlos и различные chondrodysplasias, подчеркивая критический вклад коллагеновых фибрилл в функцию тканей (Myllyharju & Kivirikko 2004). Имеются многочисленные мутантные линии мыышей, которые воспроизводят человеческие фенотипы (Aszodi et al. 1998, Myllyharju & Kivirikko 2004, Reginato & Olsen 2002). Здесь мы сфокусируемся на тех мутантных мышах, которые предоставляют информацию о сложном сценарии формирования и стабилизации фибрилл. Гетеротримерные коллагены V (α1
2α2) and XI (α1α2α3) кополимеризуются с коллагенами I и II, соотв., чтобы сформировать гетеротипические фибриллы.
In vitro эксперименты по сборке фибрилл показали. что минорные компоненты фибрилл регулируют диаметр фибрилл путем проецирования своих сохранившихся N-пропептидов на поверхность фибрилл, ограничивая тем самым добпвление новых мономеров посредством стерических и/или электростатических помех (Blaschke et al. 2000, Linsenmayer et al. 1993). В согласии с этими находками
in vitro мыши, несущие целенаправленные мутации в N-терминальных частях α2(V) цепи {
Col5α2pN/pN) обладают толстыми коллаген I фибриллами в строме роговицы (Andrikopoulos et al. 1995). Кроме того, эти мыши имеют ломкую кожу, содержащую пониженное количество рыхло упакованных фибрилл. Дальнейший анализ кожи выявил, что эти мутации тяжело нарушают внутриклеточную сборку и/или секрецию secretion of αl(V)
2α2(V) гетеротримеров и преимущественно α1(V) гомотримеры откладываются в матриксе (Chanut-Delalande et al. 2004). Эти гомотримеры. однако, не включаются в collagen I-содержащие фибриллы и формируют самостоятельные, тонкие филаменты. Как следствие инициация сборки гетеротипических фибрилл ставится под угрозу, приводя е небольшим количествам, дизорганизованных коллагеновых фибрилл в матриксе кожи. Критическая роль коллагена V в образовании гетеротипических фибрилл
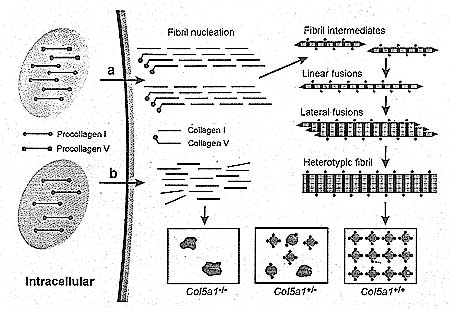
Figure 1
Regulation of fibril formation by type Y collagen, (a) In the normal situation (Col5al+/+), collagen V and collagen I copolymerize into heterotypic fibrils through a regulated fibril assembly process. Primary fibrils nucleated in the presence of collagen V form short intermediates, which subsequently grow by linear and lateral fusion events. As the retained N-terminal propeptide of collagen V reaches a certain concentration at the fibrillar surface, fibril growth may stop, and new fibril initiation becomes favorable. This mechanism leads to a population of fibrils with relatively consistent diameters, (b) When collagen V is missing owing to ocl(V) chain deficienqT(Col5a1-/-), fibril initiation is impaired, and only a few morphologically abnormal fibrils can form via unregulated self-assembly of collagen I molecules. In the case of collagen V haploinsufficiency (Col5a1+/-), fewer fibrils are nucleated and form into either normal or thick, irregular fibrils, depending on the amount of collagen V molecules incorporated into the fibrils. Excess collagen I molecules may self-aggregate to form abnormal fibrils. Adapted from Wenstrup et al. 2004.
четко продемонстрирована на мышах, лишенных коллагеновой αl(V) цепи (Figure 1) (Wenstrup et al. 2004).
Col5αl-/- животные неспособны продуцировать коллаген V и погибают на 10.5 день эмбриогенеза (El0.5) из-за сердечно-сосудистой недостаточности. Летальный фенотип сопровождается полным отсутствием collagen I-содержащих фибрилл с мезенхиме и присутствие немногих морфологически аномальных фибрилл вблизи эктодермального BM. Напротив, гетерозиготные мыши (
Col5αl+/-) выживают, но обнаруживают как нормальные, так и аномальные популяции дермальных коллагеновых фибрилл, ассоциированных с 50% снижением плотности фибрилл и коллагенового содержимого. На базе этих наблюдений, первичная роль типа V collagen заключается в контроле ранней инициации фибрилл. Коллагеновые фибриллы, инкорпорирующие collagen V, зарождаются (nucleate) и растут благодаря регулируемому процессу сборки фибрилл, тогда как aмолекулы collagen I, не ассоциированные с collagen V могут подвергаться только нерегулируемой само-агрегации, приводящей к образованию ограниченного количества морфологически аномальных фибрилл.
То что минорные коллагены являются критическими регуляторами фибриллогенеза in vivo было подтверждено с помощью collagen Xl-мутантных мышей. Естественно возникшие cho/cbo мыши содержат преждевременный стоп-кодон на N-терминальной области αl(XI) цепи, что отвечает за пониженное количество аномально толстых фибрилл в хрящевом матриксе, это приводит к сильно дизорганизованным ростовым пластинкам и к гибели при рождении с короткими конечностями и расщеплением нёба (Li et al. 1995). Мыши, лишенные Colllαl (Li et al. 2001) могут всё ещё формировать αl(XI)3 гомотримеры или гетеротримеры. состоящие из αl(XI) и α3(XI) цепей. Такие мыши обнаруживают фенотип более легкий, чем фенотип мышей cho/cho, характеризующийся формированием электрон-плотных пучков в хрящевом матриксе, указывающим на то, что α2(XI) цепь регулирует латеральные ассоциации индивидуальных коллагеновых фибрилл.
Гетеротримерный (αlα2α3) collagen IX и гомотримерный collagen XII являются структурно родственными нефибриллярными коллагенами, ассоциированными с поверхностью типа II и типа I коллагеновыми фибриллами, соотв. Мыши с целенаправленной инактивацией Col9αl гена, дают относительно легкий фенотип, характеризующийся остеоартритами с ранним началом без очевидных морфологических альтераций хрящевых фибрилл (Fassler et al. 1994) и обнаруживают прогрессирующую потерю слуха с дизорганизованной фибриллярной ультраструктурой в текториальной мембране (Asamura et al. 2005). Трансгенные мыши, экспрессирующие укороченные молекулы коллагена XII обнаруживают аномалии фибрилл только в немногих тканях, таких как periodontal ligaments и кожа (Reichenberger et al. 2000). Всё это указывает на то, что collagens IX и XII могут играть роль в организации фибриллярного коллагена и целостности ткани в специфических анатомических местах.
Elastic fibers and microfibrils. Эластические волокна, состоящие из elastin стержня и окружающей микрофибриллярной сети, обеспечивают эластичность и упругость гибких тканей. Во время эластогенеза, растворимый предшественник, молекулы tropoeiastin, откладываются в пред-сформированный микрофибриллярный матрикс и затем поперечно связываются, чтобы сформировать нерастворимые elastin полимеры. Микрофибриллы, которые также присутствуют в некоторых гибких тканях, в которых отсутствует elastin, содержат большие количества интегрированных и ассоциированных компонентов, включая fibrillin-1 и -2, fibulins, латентный transforming growth factor beta (TGF-β)-binding proteins (LTBPs) и emilins (Kielty et al. 2002). Аномалии эластических волокон ассоциируют с некоторыми патологиями, а мышиные модели, которые помогают нам понять болезни эластических тканей у людей, получены в последнее время.
У людей гаплонедостаточность elastin вызывает supravalvular aortic stenosis (SVAS), обструктивное сосудистое нарушение, характеризующееся сужением крупных артерий. Как гетерозиготные мыши, несущие нулевую мутацию гена elastin (Eln+/-), так и у SVAS пациентов обнаруживается утолщение стенки аорты, связанное с повышенным количеством эластиновых lamellae и сосудистых smooth muscle cells (SMCs), это указывает на то, что понижение уровня elastin индуцирует механизм онтогенетической компенсации, который приводит к образованию множества эластических колец (Li et al. 1998b). Elastin-null мыши {Eln-/-) обнаруживают нормальное развитие артерий во время эмбриогенеза: однако, они вскоре погибают после рождения из-за закупорки артерий, вызываемой избыточной пролиферацией и субэндотелиальным накоплением сосудистых SMCs (Li et al. 1998a). Следовательно, elastin безусловно важен и для позднего артериального морфогенеза, регулируя пролиферацию, миграцию и организацию сосудистых SMCs (Karnik et al. 2003, Li et al. 1998a).
Fibrillin-1 и -2 являются cysteine-богатыми гликопротеинами, которые полимеризуются в структуры, напоминающие бусинки на тесемке, а после латеральной ассоциации индивидуальных полимеров, создаётся структурная основа микрофибриллярной решетки (Ramirez et al. 2004). У людей мутации fibrillin-1 приводят к синдрому Marfan (MFS), плейотропному нарушению сердечно-сосудистыми, глазными и скелетными аномалиями. Сердечно-сосудистые осложнения являются основным источником смертности и болезненности при MFS, включая аневризмы аорты и рассечения, а также пролапс митрального клапана (Milewicz et al. 2000). Мутации в гене fibrillin-2 человека вызывают врожденную контрактурную arachnodactyly, MFS-родственное нарушение с преимущественно проявлениями в скелетных мышцах (Milewicz et al. 2000). Мышиные модели, несущие hypomorphic (т.e., вызывающие пониженную экспрессию белка) или доминантно-негативные мутации fibrillin-1, вызывают разрывы аорты на разных стадиях постнатального развития и воспроизводят ранние и поздние летальные формы MFS, указывая тем самым, что дозовые эффекты или природа мутаций fibrillin-1 влияет на тяжесть MFS (Pereira et al. 1997, 1999; Judge et al. 2004). Неожиданно эти мыши обнаруживают нормальную морфологию эластичных волокон в неповрежденных областях аорты и в незатронутых тканях, указывая, что первичная роль fibrillin-1 заключается в поддержании целостности ткани (Ramirez et al. 2004). Однако, это мнение было поставлено под сомнение, т.к. мыши, лишенные fibrillin-1 (Fbn1-/-)? обнаруживают нарушенное созревание неонатальной стенки аорты и погибают приблизительно спустя 14 дней после рождения от разрыва подверженных механическим стрессам эластических тканей (Carta et al. 2006). Мыши, лишенные fibrillin-2 {Fbn2-/-) жизнеспособны и без каких-либо признаков сосудистых дефектов, но обнаруживают синдактилию из-за дефектной передачи сигналов bone morphogenetic protein (BMP) (Arteaga-Solis et al. 2001). С др. стороны, fibrillin-1/fibrillin-2 двойные нехватки вызывают эмбриональную летальность, ассоциированную с задержкой/нарушением эластогенеза (Carta et al. 2006). Следовательно, fibrillin-1 и -2 выполняют перекрывающиеся функции в ранней сборке эластичных волокон; более того, fibrillin-1 выполняет двойную роль в регуляции развития стенки аорты и в поддержке структурной целостности force-bearing эластических тканей.
Fibulin-5 (Fbl-5) является модулярным гликопротеином, который связывает эластичные волокна с клетками путем связывания как tropoelastin, так и integrins (Nakamura et al. 2002, Yanagisawa et al. 2002). Функция образования таких мостиков предсказывает роль Fbl-5 в эластогенезе. В самом деле, Fbl5-/- мыши, обладающие дизорганизованными и фрагментированными эластичными волокнами, получают эмфизему легких, дряблую кожу и сосудистые аномалии (Nakamura et al. 2002, Yanagisawa et al. 2002). Такой фенотип напоминает клинические проявления тяжелой аутосомно-рецессивной формы cutis laxa, нарушения, которое характеризуется дряблой кожей и легочной эмфиземой и вызывается гомозиготными missense мутациями в гене FBL5 (Loeys et al. 2002).
LTBPs 1-4 являются родственными fibrillin молекулами с двумя предполагаемыми функциями: в качестве структурных компонентов ECM и в качестве модуляторов доступности TGF-β (Hyytiainen et al. 2004). Среди мышиных моделей для LTBPs, только LTBP-4 гипоморфные мыши обладают четким структурным дефектом. Эти мыши характеризуются легочной эмфиземой и ректальным prolapse, ассоциированными с фрагментированными эластическими волокнами (Sterner-Kock et al. 2002), подтверждая, что LTBP-4 играет роль в развитии эластичных волокон посредством взаимодействия с микрофибриллами в специфических тканях.
Emilin-1, который принадлежит к семейству emilin/multimerin белков ECM, локализуется на интерфейсе эластинового стержня и микрофибрилл. Elastin, fibulin-5 (Zanetti et al. 2004) и β1 integrin (Spessotto et al. 2003) соединяются с emilin-1, указывая тем самым, что emilin-1выполняет множественные роли в эластогенезе, в поддержании собственно организации эластичных волокон и в зависимых от закрепления клеточных функциях. Emilin-1 нокаутные мыши имеют нормальную продолжительность жизни, но обнаруживают некоторые средней тяжести альтерации в коже и аорте, включая аномальные эластичные волокна, уменьшение соединений клетка-эластичные ламеллы и морфологические дефекты клеток (Zanetti et al. 2004).
Proteoglycans in tissue stability. PGs состоят из стержневого белка, ковалентно связанного с боковыми цепочками glycosaminoglycan (GAG). Gly-cosaminoglycans являются сульфатированными олигосахаридами, состоящими из повторящихся дисахаридных единиц из dermatan sulfate (DS), heparan sulfate (HS)/heparin, chondroitin sulfate (CS) или keratan sulfate (KS). PGs играют важную роль в гидратационных и осмотических свойствах матрикса, но как предполагается обладают некоторыми дополнительными функциями благодаря взаимодействиям с различными компонентами матрикса, факторами роста и рецепторами клеточной поверхности.
Hyalectans являются крупными CS-содержащими PGs, которые образуют агрегаты с hyaluronan, не сульфатированным GAG полимером, дают повторяющиеся дисахаридные единицы. Семейство состоит из 4-х членов с разными тканевыми распредениями: Versican является наиболее широко распространенным hyalectan, a aggrecan находится преимущественно в хрящах, тогда как neurocan и brevican ограничены головным мозгом, хотя все hyalectans, как полагают, являются важными дл организации DRV? эксперименты с нокаутами подтверждают эжту гипотезу только для aggrecan и versican. Мыши, гомозиготные по инсерционной мутации в гене Gspg2, кодирующем versican, погибают приблизительно на El0.5 из-за тяжелых проблем с сегментацией сердца, связанных с несоответствующим разбухание матрикса эндокардиальных подушек и из-за неспособности клеточной миграции (Aljaatvedt et al. 1998). Мыши, лишенные hyaluronan synthase 2, дают сходный фенотип (Camenisch et al. 2000), указывая тем самым, что взаимодействие versican-hyaluronan является существенным для генерации условий в DRV? пригодных для клеточной миграции во время развития сердца. В хрящах hydrated природа aggrecan-hyaluronan агрегатов предоставляет ткани упругость и резистентность против сил сжатия. Отсутствие aggrecan у спонтанных мутантов audi и у линий мышей ведет к перинатальной летальности7 благодаря расщеплению нёба и тяжелым нарушениям образования эндохондральных костей; такие нарушения характеризуются компрессией хрящевого ВКМ, дизорганизацией ростовых пластинок и альтерациями в паттерне экспрессии специфичных для хрящей генов (Wai et al. 19,98, Watanabe et al. 1994). По сравнению с aggrecan- и versican-нулевыми мутаций, мыши, лишенные как neurocan, так и brevican жизнеспособны и не обнаруживают морфологическийх алтераций в ЦНС (Rauch et al. 2005). Однако, нехватка neurocan или brevican нарушает долговременную потенциацию гиппокампа, указывая на роль этих hyalectans в синаптической пластичности (Brakebusch et al. 2002, Zhou et al. 2001).
Семейство small leucine-rich proteoglycans (SLRPs) как полагают сегодня состоит из 13 членов. SLRPs характеризуются относительно небольшим стрежневым белком, содержащим варьирующие количества leucine-rich repeat (LRR) единиц, которые фланкированы связанными дисульфидными мостиками, богатыми цистеином доменами на N и C концах (Ameye & Young 2002). SLRPs, такие как decorin, biglycan и fibro-modulin широко распределены в теле, тогда как др. подобно lumican, keratocan или mimecan обнаруживают более ограниченный паттерн экспрессии. Некоторые SLRPs соединяются с фибриллярными коллагенами посредством LRR домена и регулируют сборку коллагеновых фибрилл in vitro. Decorin, наиболее изученный SLRP, соединяется посредством своих LLRs вблизи С конца индивидуальных коллагеновых фибрилл, регулируя боковое слияние и поддерживает также межфибриллярные пространства посредством своей DS боковой цепи (Reed & Iozzo 2002). Распределение SLRPs на коллагеновых фибриллах, их GAG половинка7, их дисульфидные петли и их потенциал мультимеризации также считаются важными для боковой сборки, ориентации и стабильности фибрилл (Waddington et al. 2003). Мыши, лишенные членов этого семейства имеют типа I collagen-содержащие фибриллы с нерегулярным диаметром и/или организацией и имеют широкий круг болезней (e.g., osteoporosis, osteoarthritis, Ehlers-Danlos syndrome и различные болезни роговицы) благодаря измененной механике или структуре ткани (Ameye & Young 2002, Kao & Liu 2002). Хотя in vivo фенотипы разных SLRP нокаутов подтверждают значение этих PGs в коллагеновом фибриллогенезе, понимание механизмов еще далеко от завершения.
Perlecan является heparan sulfate proteoglycan (HSPG) с широким паттерном экспрессии. Стержневой белок perlecan состоит из 5 самостоятельных доменов: трех HS цепей, прикрепленных к N-терминальному домену I и дополнительная цепочка потенциально локализуемая в домене V. Perlecan-нулевые (
Hspg2-/-) мыши погибают на двух стадиях: на E10.5-12.5 из-за BM дефектов и перинатально из-за респираторной неспособности (Arikawa-Hirasawa et al. 1999, Costell et al. 1999). Мутанты, которые погибают при рождении являются карликовыми и обнаруживают тяжелые дефекты хрящей. Ростовая пластинка дизорганизована, а пролиферация и дифференцировка хондроцитов, а также последующее образование эндохондральной кости нарушены (Arikawa-Hirasawa et al. 1999, Costell et al. 1999). Эти дефекты могут возникать из-за структурно измененного ВКМ, т.к. мутантные хрящи содержат меньше и более короткие коллагеновые фибриллы (Costell et al. 1999). Механизм. лежащий в основе снижения плотности коллагена
7, неясен, но perlecan может помочь защитить коллагеновую сеть от деградации путем связывания и регулирования активности matrix metalloproteinases (ALMPs) (Costell et al. 1999). Хрящевые аномалии у
Hspg2-/- мышей напоминают те, что наблюдаются при Silver-Handmaker type dyssegmental dysplasia, летальном аутосомнощрецессивном синдроме у людей, вызываемом нулевыми мутациями в гене perlecan (Arikawa-Hirasawa et al. 2001).
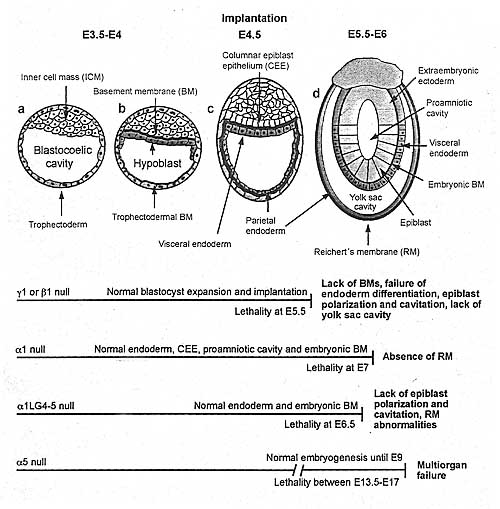
Рис.2.
|
The role of laminins in peri-implantation development. The fully expanded, zona-free blastocyst contains two distinct components: the inner cell mass (ICM) and the trophectoderm shell. Prior to implantation, ICM cells lining the blastocoelic cavity differentiate into hypoblast (or primitive endoderm) cells, which deposit a basement membrane (BM) between them and the remaining undifferentiated ICM. This BM is continuous with the BM deposited by the trophectoderm. After implantation at E4.5, the hypoblast gives rise to two extraembryonic cell lineages: visceral endoderm cells that differentiate from hypoblast cells in contact with the BM and parietal endoderm cells that migrate along the trophectodermal BM. ICM cells adjacent to the BM become polarized and develop into columnar epiblast epithelium (CEE), whereas interior ICM cells undergo apoptosis. By E5.5-E6, the proamniotic cavity forms, and the visceral endoderm and the epiblast (or embryonic ectoderm) are separated by the embryonic BM. Parietal endoderm cells secrete BM components, which incorporate into Reichert's membrane (RM). During peri-implantation, laminin-111 (αlβlγl) and laminin-511 (α5βlγl) are expressed; the consequences of chain-specific gene mutations in mice are described {bottom). The Role of ECM Components in Basement Membrane Integrity
BMs состоят преимущественно из collagen IV, laminins, nidogens и perlecan. Дополнительными компонентами из зон BM являются agrin, fibulins, fibronectin (Fn) и различные нефибриллярные коллагены. Тонкая структурная архитектура и репертуар взаимодействий среди структурных белков и молекул клеточной поверхности в BM зонах может варьировать для выполнения ткане-специфических потребностей (Miner et al. 2004 and Yurchenco et al. 2004).
Collagens modulate the integrity and function of basement membrane zones. Наиболее богатым коллагенозным компонентом BMs является сеть, формируемая коллагеном типа IV, который состоит из гетеротримерной комбинации 6 самостоятельных полипептидных цепочек. Хотя главная изоформа эмбрионального collagen IV, α12α2(IV), экспрессируется уже на ст. бластоциста, мыши с целенаправленной инактивацией локуса Col4al/Col4a2 (Poschl et al. 2004) продолжают развиваться вплоть до E9.5 и откладывают BM-подобные слои в отсутствие какой-либо из изоформ collagen IV. После E9.5 дефицит αl2α2(IV) ведет к нестабильности BM, приводящей к разрыву подверженных механическим стрессам BMs и Reichert's membrane (RM)- специализированной BM, которая отделяет эмбрион от материнских тканей - это приводит к гибели между E10.5-E11.5. Мыши, несущие доминантно-негативную мутацию в гене Col4al, которая предупреждает секрецию гетеротримеров collagen IV, также формируют рвущиеся BMs и погибают в средине беременности (Gould et al.2005). Гетерозиготные мыши, имеют пониженную перинатальную жизнеспособность и церебральные кровотечения, а некоторые выживающие бладают porencephaly-подобным фенотипом. Porencephaly редкое нейрологическое заболевание, характеризующееся церебральными повреждениями и дегенеративными полостями, возникающими из-за кровотечений, что приводит к недееспособности и гибели в некоторых случаях. Collagen IV может находиться среди факторов предрасположенности для porencephaly, т.к. мутации в гене COL4A1, были найдены в двух затронутых семьях (Gould et al. 2005). Всё это указывает на то, что сеть collagen IV не существенна для сборки BM, но необходима для поддержания её целостности в условиях механических нагрузок.
Мыши. несущие мутации в др. коллагенах BM-зоны, дают фенотипические отклонения в разных анатомических местах, указывая на специализированные роли эти х коллагенов (rev. Gustafsson & Fassler 2000). Напр., мыши, лишенные в количественном отношении минорного α3 или α3/α4 цепочек collagen IV, обнаруживают glomerulonephritis, recapitulating autosomal forms of Alport syndrome, в то время как дефицит collagen VII ведет к кожным волдырям, напоминающим dystrophic epidermolysis bullosa у людей.
Такой анали з различных трансгенных линий мышей выявил особенно важную роль коллагенов BM-зоны в развитии мышц и глаз. Collagen VI похож на филаменты-формирующий коллаген, состоящий из трех самостоятельных цепочек. Было предположено, что он играет роль в прикреплении BM неэпителиальных клеток к подлежащему матриксу, закрепляя сеть collagen IV на компонентах интерстициума. interstitium. Col6al-нулевые мыши обнаруживают мышечные аномалии, такие как нерегулярный диаметр волокон, некроз волокон и снижение контрактильной силы мышц (Bonaldo et al. 1998, Irwin et al. 2003). Фенотип, напоминающий Bethlem myopathy, мышечное нарушение с ранним началом и с аутосомно-доминантным наследованием, ассоциирует с мутациями в генах, кодирующих collagen VI (Lampe & Bushby, 2005).
Collagen types XV и XVIII являются примерами почти везде экспрессирующихся, высокогликозилированных коллагенов BM-зоны с ткане-ограничивающей функцией. Мыши, лишенные коллагена XV, жизнеспособны, но страдают от прогрессирующей скелетной миопатии и сердечно-сосцдистых дефектов (Eklund et al. 2001). BMs выглядят нормальными у мутантов, указывая тем самым, что collagen XV не нужен для сборки BM, но он необходим для осуществления сцепления между клетками и окружающим ВКМ для поддержания механической стабильности мышц и капилляров. Дефицит коллагена XVIII у мышей ведет к различным дефектам глаз, сопровождающих аномалии BM (rev. Marneros & Olsen 2005). Дефекты включают снижение прикрепления коллагеновых фибрилл стекловидного тела к BМ, покрывающей поверхность сетчатки; разрывы радужки; атрофию цилиарных тел: зависимая от возраста потеря зрения с нарушениями функции пигментного эпителия сетчатки. Большинство из этих аномалий обнаруживается у пациентов с синдромом Knobloch, аутосомно-рецессивным нарушением, вызываемым инактивирующими мутациями в гене COL18A1 человека (Suzuki et al. 2002). Дефектные BMs у Coll8al-/- мышей, однако, не ограничены глазами: утолщенные BMs обнаруживаются в хороидном сплетении, что ведет к дилятации мозговых желудочков и к увеличению зависимой от генетического фона чувствительности к гидроцефалии (Utriainen et al. 2004). Т.о., коллаген XVIII действительно играет роль в структуре BM и затрагивает некоторые эпителиальные функции, регулируя взаимодействия между клетками и матриксом.
Basement membrane glycoproteins: the laminin networks. Laminins являются семейством из 16 гетеротрехмерных (αβγ) гликопротеинов, генерируемых комбинацией из 5α, 4β и 3γ wtgtq. Полимерные ламининовые сети само-ассоциируют, чтобы взаимодействовать с ВКМ и молекулами клеточной поверхности, такими как nidogen, perlecan, integrins и α-dystroglycan. Dct BMs содержат laminins, но распределение изоформ регулируется онтогенетическим и ткане-специфическим образом (Miner & Yurchenco 2004, Yurchenco et al. 2004). Здесь мы будем обозначать ламинины согласно новой упрощенной номенклатуре, в которой численное обозначение ламининов арабскими цифрами соответствует α β и γ цепям. соотв. (Aumailley et al. 2005).
Laminin-111 и laminin-511 являются двумя изоформами ламинина, присутствующми в синтезируемых BMs из пери-имплантационных эмбрионов (Figure 2). Мыши, дефицитные по β1 или γl субхединицам, погибают вскоре после имплантации (на ст. E5.5), не обнаруживая ламининовых тримеров и лишенные как эмбриональной BM (которая отделяет эпибласт от висцеральной энтодермы), так и Reichert's мембраны, подтверждая тем самым, что ламинины являются ключевыми молекулярными компонентами, инициирующими сборку BM (Miner et al. 2004, Smyth et al. 1999). Отсутствие эмбриональных, исходящих из BM сигналов нарушает дифференцировку энтодермы, поляризацию эпибласта и образование проамниотической полости. Напротив, αl- или α5-нулевые мыши доживают вплоть до E7 и E13.5-E17, соотв., указывая тем самым. что laminin-111 и laminin-511 могут частично компенсировать др. др. во время раннего развития (Miner & Li 2000; Miner et al. 1998, 2004). Напр., у αl-дефицитных мышей эмбриональная BM и проамниотическая полось образуются, тогда как RM отсутствует (Miner et al. 2004). Более того, хотя избыточная экспрессия трансгенной α5 субъединицы у αl-нулевых мышей удлиняет жизнеспособность до E7.5 и делает возможной инициацию гаструляции, она не поддерживает образование RM (Miner et al. 2004). Эти исследования показали критическую роль laminin-111 в образовании Reichert's мембраны, но остается вопрос, может ли эмбриональная BM, содержащая только laminin-511 поддерживать развитие, если RM оказывается незатронутой. Интересно, что мыши, экспрессирующие структурные мутации αl цепи, лишенной последних двух C-терминальных laminin globular (LG4-5) доменов - которые обеспечивают взаимодействия с dystroglycan, heparin и sufatides - погибают на полдня раньше, чем αl-нулевые животные (Scheele et al. 2005). Несмотря на сборку эмбриональной BM, цилиндрический эпителий эпибласта и проамниотическая полость не образуются у α1LG4-5-/- мышей, указывая, что αl цепь индуцирует дифференцировку эпибласта посредством своих LG4-5 доменов. Т.к. эпибласт является поляризованным у αl-нулевых мышей, то эти данные также указывают на то, что экспрессия укороченной αl цепи предупреждать компенсаторную позитивную регуляцию α5 цепи в эмбриональной BM. Более того RM собирается, но фрагментирована у большинства αlLG4-5-/- мышей, демонстрируя, что всяα1 цепь необходима для полной структурной стабильности RM.
После раннего эмбриогенеза BMs экспрессируют различные субнаборы изоформ laminin ткане-специфическим способом, чтобы выполнить функциональное предназначение в развивающемся органе. Важность ламининов для функции BM во время позднего эмбрионального и постэмбрионального развития продемонстрирована при анализе линий м sitq с естественно возникшими и целенаправленно индуцированными мутациями в генах, кодирующих субъединицы ламининов (rev. Miner & Yurchenco 2004, Yurchenco et al. 2004). Напр., дефицит laminin α2 вызывает тяжелую мышечную дистрофию и дефекты периферических нервов; устранение гена, кодирующего laminin α3 вызывает кожную пузырчатку; отсутствие α4 цепи ведет к мягкой мышечной дистрофии и нарушению созревания микрососудов; а отсутствие laminin α5 приводит к аномалиям конечностей, почек, плацентарных сосудов и нервной трубки, а также к дефектам дифференцировки интерстициальных гладких мышц. Хотя в некоторых случаях описывается компенсаторная позитивная регуляция др. ламининов, это обычно не восстанавливает фенотипа полностью.
Члены nidogen семейства гликопротеинов, nidogen-1 и -2, являются повсеместно экспрессируемыми компонентами эмбриональных BMs с широким кругом партнеров по связыванию. Они, как полагают, являются центральными организаторами BMs, связывающими ламининовую и колагеновую сети и включающие др. молекулы ВКМ в базовый каркас (Yurchenco et al 2004). В противоположность предполагаемой роли nidogens в сборке BM, и nidogen-1- и nidogen-2-нулевые мыши (Nidl-/- и Nid2-/-) здоровы и не имеют явных дефектов BM defects (Murshed et al. 2000, Schymeinsky et al. 2002), хотя некоторые разрывы BM капилляров головного мозга сопровождаются легкими нейрологическими нарушениями у Nidl-/- мышей (Dong et al. 2002). Неожиданно даже у
Nid1-/-/Nid2-/--двойных нокаутных мышей (Bader et al. 2005) и мышей, лишенных nidogen-связывающего сайта в γ1 цепи ламинина (Willem et al. 2002), имеются BMs, указывающие на то, что ни nidogens, ни nidogen-laminin взаимодействия per se не являются критическими для сборки BM. Однако, эти мыши погибают вскоре после рождения от респираторного distress из-за задержанного созревания легких. Nidogen-1 и nidogen-2 двойная недостаточность также приводит к к ультраструктурным аномалиям кардиальных BMs, указывая тем самым, что nidogens специфически необходимы для поддержания определенных BMs и для определенных процессов органогенеза.
The roles of perlecan and agrin. В BMs, наиболее многочисленным PG является perlecan, который соединяется с широким кругом молекул, включая белки BM-зоны (напр., laminin, collagen IV, entactin/nidogen, fibulin, collagen XVIII/endostatin), рецепторы клеточной поверхности (напр., integrins, α-dystroglycan) и ростовые факторы [напр., fibroblast growth factors (FGFs), vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF)] (rev. Iozzo 2005). Хотя репертуар этих взаимодействий подтверждает, что perlecan является важным для сборки BM, Hspg2-/- мыши формируют эмбриональные BMs. Однако, разрушения BMs в областях с повышенными механическими нагрузками, в таких как сокращающийся миокард, ведут к тканевым повреждениям и кровотечениям в перикардиальную полость и к гибели большинства мутантов между E10-E12.5 (Costell et al. 1999). Эти находки демонстрируют, что perlecan несущественнен для образования BM, но он критически необходим подобно collagen IV для поддержания целостности BM под воздействием механических усилий. Интересно, что Hspg2-/- мыши, которые доживают вплоть до рождения, имеют пониженные количества богатых fibrillin микрофибрилл в эпидермально-дермальных соединениях кожи, указывая тем самым, что perlecan является важным для биогенеза микрофибрилл и/или прикрепления этих структур к BM (Tiedemann et al. 2005).
В neuromuscular junctions (NMJs), perlecan образует кластеры в постсинаптической BM с уникальным набором молекул, включая acetylcholinesterase (AChE), acetylcholine (ACh) receptors (AChRs), agrin, и dystroglycans (Bezakova & Ruegg 2003). У Hspg2-/- мышей, AChE синтезируется, но полностью исключен из but completely excluded from NMJs, показывая, что perlecan является важным для локализации AChE в синаптической BM (Arikawa-Hirasawa ct al. 2002). Это согласуется с пониженными количествами кластеров AChE у пациентов с синдромом Schwartz-Jampel, легкой chondrodystrophies myotonia, вызываемой мутациями в гене perlecan, дающими укороченные белковые формы (Arikawa-Hirasawa et al. 2002).
Мыши, экспрессирующие perlecan, который лишен мест прикрепления к трем HS цепочкам домена I (Hspg2δ3/δ3), не обнаруживают крупных дефектов, указывая тем самым, что эти HS цнпочки не важны для целостности большинства BMs и хряща. Однако, Hspg2δ3/δ3 мыши имеют маленькие глаза и у них дегенерируют хрусталики в течение 3-х недель после рождения из-за структурных аномалий капсулы хрусталика (Rossi et al. 2003).
В то время как эксперименты с трансгенами указывают на широкий круг in vivo функций perlecan, agrin в основном известен как критический игрок в постсинаптической дифференцировке NMJs. Альтернативный сплайсинг дает ряд ткане-специфичных agrins: так наз. - z+-agrin экспрессируется нейрональными клетками и дает кластеры AChRs, тогда как z--agrin экспрессинуется не-рнейрональными клетками и неэффективно индуцирует аггрегаты AChR (rev. Bezakova & Ruegg 2003). Мыши, лишенные agrin (agrn-/-) or z+-agrin, погибают при рождении из-за респираторного distress, обнаруживают заметное снижение кластеров AChR и неспособность формировать функциональные постсинаптические структуры (Burgess et al. 1999, Gautam et al. 1996). Дальнейшие генетические исследования показали, что agrin участвует в росте и стабилизации постсинаптических структур, но не нужен для инициации образования кластеров AChR (Lin et al. 2001, Yang et al. 2001). Интересно, что делеция гена, кодирующего Ach-synthesizing enzyme choline acetyltransferase (chat-/-), на agrin-нулевом фоне почти полностью устраняет дефекты образования кластеров AChR и созревания NMJ (Alisgeld et al. 2005). In vitro эксперименты с культивируемыми миобластами показали, что ACh действует в качестве AChR-declustering фактора, в то время как agrin действует как antideclustering молекула, защищающая кластеры от индуцируемой ACh дестабилизации. Т.о., in vivo agrin не только играет роль в образовании кластеров AChR, но и также важен для противодействия диспергирующему эффекту ACh. Роль agrin вне или в NMJs менее ясна. Agrin-дефицитные эмбрионы обнаруживают менее дифференцированные, межнейрональные симпатические синапсы и дефектную синаптическую передачу в верхнем цервикальном ганглии, указывая, что нейрональный agrin координирует синаптогенез по всей нервной системе (Gingras et al. 2002).
Недавно изоформа z
--agrin была клинически увязана с коренным улучшением при laminin oil-deficient congenital muscular dystrophy (MDCA1). MDCA1 является болезнью мышечной атрофии, которая часто ведет к гибели в раннем детском возрасте из-за отсутствия BM компонента laminin-211, который соединяет мышечную BM с плазматической мембраной посредством dystroglvcans и integrins. У больных индивидов вместо него откладывается laminin-411; однако, он не может связывать dystroglycan. Избыточная экспрессия миниатюрной формы z
--agrin, которая может поперечно связывать laminin α4 цепь с dystroglycan, восстанавливает целостность BM и улучшает функцию мышц в этой животной модели (Bentzinger et al. 2005, Moll et al. 2001, Qiao et al. 2005).
ECM COMPONENTS AND SIGNALING
Рассмотренные выше нокаутные мыши демонстрируют как компоненты ВКМ вносят вклад в каркас матрикса или в качестве базовых структурных элементов или в качестве организаторов/модуляторов структурной сети. Однако, ВКМ генерируют также инструктивные сигналы, влияющие на клеточное поведение путем обеспечения взаимодействий между клетками и матриксом и путем регуляции доступности или презентации ростовых факторов. Растут доказательства, указывающие, что все классы матричных молекул участвуют в индукции сигнальных процессов.
Collagens and Signaling
До последнего времени коллагены рассматривались как структурные компоненты ВКМ. Недавний анализ трансгенных мышей, обладающих мутантными коллагеновыми генами, предоставил доказательства, что это слишком упрощенная точка зрения. Col6al-/- мыши, напр., демонстрируют неожиданную дисфункцию мышечных митохондрий (Irwin et al. 2003). Collagen Vl-дефицитные мышцы обнаруживают высокие уровни спонтанного апоптоза и ультраструктурные аномалии саркоплазматического ретикулема и митохондрий. Разнообразные стимулы, такие как оксидативные стрессы или перегрузка Ca2+ могут открывать высокую проводимость permeability transition pores (PTPs) во внутренней мембране митохондрий, приводя к деполяризации и разбуханию митохондрий, которые в свою очередь инициируют пути, ведущие к апоптозу и некрозу (Rizzuto 2003). Обработка collagen VI-дефицитных мышей cyclosporin A (CsA) (aблокирует открытие PTP) устраняет ультраструктурные дефекты и снижает апоптоз. Следовательно, возможно лечить связанные с collagen VI мышечные нарушения, применяя просто CsA. Однако, механизм. который связывает дефицит collagen VI с аномалиями митохондрий всё ещё неизвестен. Т.к. collagenous домен коллагена VI взаимодействует рецепторами клеточной поверхности интегринов, то отсутствие такого взаимодействия скорее всего затрагивает зависимые от интегринов пути клеточной жизнеспособности.
Collagen XIX является гомотримерным, редким коллагеном BM-зоны с преимущественной экспрессией в дифференцирующихся мышечных клетках во время эмбриогенеза. Большинство мышей, лишенных коллагена XIX (Coll9alN/N), погибает в течение первого мес. постнатальной жизни, тогда как мыши, откладывающие структурно аномальные молекулы (Coll9alδ/δ) выживают, но имеют ослабленную приспособляемость на поздних взрослых стадиях (Sumiyoshi et al. 2004). И Coll9alN/N и Coll9alδ/δ мыши также страдают от гипертензивности lower esophageal sphincter (LES), обусловленной нарушением, индуцируемой глотанием nitric oxide (NO)-зависимой реляксации гладкомышечных клеток в этой области. BM, окружающая SMCs у мутантных животных, является аномальной, указывая на то, что транспорт NO от nitrergic нервов к SMCs критически зависит от соотв. организованного матрикса, включая XIX. SMC дисфункция и неспособность к реляксации LES являются типичными признаками у пациентов с achalasia (Goyal 2001), моторным нарушением пищевода в основном с неизвестной генетикой. Achalasia-подобный фенотип у Col19a1-мутантных мышей открывает возможность, что collagen XIX участвует в этиологии этого заболевания у человека. Кроме того, collagen XIX играет существенную роль в трансдифференцировке мышц абдоминального сегмента пищевода (Sumiyoshi et al. 2004). У нормальных мышей наружный мышечный слой пищевода подвергается smooth-to-skeletal-muscle превращению в кранио-каудальном направлении вплоть до 3-й недели постнатальной жизни. Эта программа трансдифференцировки находится под контролем myogenic regulatory factors (MRFs), таких как MyoD или myogenin, которые индуцируют транскрипцию генов. специфичных для скелетных мышц. У мышей дикого типа экспрессия myogenin постепенно прогрессирует от уровня диафрагмы в абдоминальном сегменте пищевода, но у Col19a1-/- мышей экспрессия myogenin останавливается на уровне диафрагмы и трансдифференцировка дальше не идет. Эти данные указывают на участие collagen XIX в контроле распределения активности сигнальных молекул ВКМ, которые запускают экспрессию гена MRF в нижней части пищевода. Альтернативно collagen XIX может модулировать взаимодействия между клетками и матриксом, которые существенны для скелетного миогенеза.
Тройная спиральная структура нативных фибриллярных коллагенов соединяется с и активирует рецепторную тирозин киназу DDR2 (discoidin domain receptor 2), которая затем индуцирует экспрессию MMPs (rev. Tran et al. 2005). Мыши, гетерозиготные по мутации collagen XI (
cho/+), обнаруживают остеоартрит-подобные изменения в своих коленных суставах (Xu et al. 2003), что сопровождается возраст-зависимой совпадающей индукцией экспрессии DDR2 и MMP-13 (Xu et al. 2005). Эксперименты с культивируемыми нормальными хондроцитами показали, что активация DDR2 с помощью коллагена типа II ведет к позитивной регуляции MMP-13 (Xu et al. 2005). Считается, что
Col11a1 гаплонедостаточность изменяет распределение коллагеновой II/XI фибриллярной сети у
cho/+ мышей, приводя к увеличению количества взаимодействий между мутантными фибриллами и DDR2. Индукция активности MMP-13 с помощью передачи сигналов DDR2 существенно увеличивает деградацию collagen II в мутантном суставном хряще, характерный признак прогрессирующего остеоартрита.
Proteoglycans and Growth Factor Signaling
PGs могут участвовать с молекулярных взаимодействиях с секретируемыми сигнальными молекулами и рецепторами клеточной поверхности. Среди внеклеточных HSPGs, perlecan действует как акцессорный рецептор низкого сродства для FGF-2 и обеспечивает FGF-зависимую пролиферацию клеток и ангиогенез (Aviezer et al. 1994). Получены генетические доказательства ангиогенез-модулирующей роли perlecan, Hspg2δ3/δ3 мыши обнаруживают нарушение индуцируемой FGF-2 неоваскуляризации, задержку заживления ран и задержку роста опухолей (Zhou et al. 2004). Эти данные указывают на то, что perlecan регулирует передачу сигналов FGF-2 и ангиогенез посредством своих HS боковых цепочек. Также нарушенная передача сигналов FGF в ростовой пластинке, как предполагается частично ответственна за дефект пролиферации хондроцитов у Hspg2-/- мышей (Arikawa-Hirasawa et al. 1999). Напротив perlecan-нулевые эмбрионы обнаруживают гиперплазию SMC-специфических мезенхимных клеток в тракте оттока сердца, что приводит к врожденным порокам крупных и коронарных артерий сердца (Costell et al. 2002, Gonzalez-Iriarte et al. 2003). Perlecan ингибирует пролиферацию SMC посредством опухолевого супрессора PTEN, фосфатазы. которая негативно регулирует Akt (protein kinase B) и focal adhesion kinase signaling (Garl et al. 2004). Как perlecan влияет на активность PTEN неизвестно, но взаимодействия между стержневым белком и рецепторами клеточной поверхности, как полагают. регулируют активность PTEN. Альтернативно HS цепочки perlecan могут секвестрировать ростовые факторы, снижая тем самым обеспечиваемую ростовыми факторами пролиферацию SMC. В подтверждение этой гипотезы, Hspg2δ3/δ3 мыши обнаруживают усиленный рост SMC и гиперплазию интимы после повреждения каротидной артерии (Tran et al. 2004).
Нокаутные модельные мыши предоставляют доказательства роли SLRPs в передаче сигналов ростовых факторов и непосредственно регуляции роста клеток. По крайней мере, три SLRPs (decorin, fibromodulin и biglycan) взаимодействуют с TGF-β, цитокином, участвующим в таких клеточных процессах. как пролиферация, жизнеспособность и дифференцировка(Ameye & Young 2002). У biglycan-нулевых и biglycan/decorin двойных нокаутных мышей наблюдается остеопения, ассоциированная с повышенным апоптозом bone marrow stromal cells (BMSCs), приводящие к снижению способности BMSCs формировать колонии in vitro (Bi et al. 2005, Chen et al. 2002). Это указывает на то, что пониженные количества osteoprogenitor клеток частично ответственны за костные дефекты у этих мышей. Одновременное отсутствие biglycan и decorin в стромальном матриксе приводит к повышению доступа к TGF-β1, приводя к избыточной активации TGF-β1-обеспечиваемых сигнальных путей (Bi et al. 2005). Biglycan/decorin-дефицитные BMSCs обнаруживают достоверно более высокий уровень фосфорилирования чувствительных к TGF-β нижестоящих сигнальных молекул Smad2 и Smad3, а также к повышению активности ключевой апоптической протеазы caspase-3. Активность Smadl, которая обеспечивает передачу сигналов от др. членов TGF-β сверхсемейства, таких как BMP2 и BMP4, также увеличена в двойных мутантных клетках. Из-за нормальной способности к дифференцировке мутантных BMSCs можно предполагать, что biglycan и decorin кооперируются, чтобы модулировать баланс между ростом и апоптозом BMSCs, контролируя количества секвестрированных и свободных TGF-β в матриксе костного мозга. Напротив, когда отсутствует только biglycan, то дифференцировка пре-остеобластов свода черепа в зрелые остеобласты нарушена из-за снижения BMP4-обеспечиваемой передачи сигналов (Chen et al. 2004). В предлагаемой модели decorin и biglycan могут конкурентно связывать BMPs/TGF-β, чтобы секвестрировать их в матриксе или предоставить их клеточным рецепторам, соотв. Т.о., в отсутствие biglycan в присутствии decorin эти ростовые факторы закрепляются в матричном компартменте, понижая доступные количества BMPs. В отсутствие и biglycan и decorin, BMSCs получают неограниченный доступ к ростовым факторам. Всё это указывает на то, что decorin и biglycan играют важную роль в предопределении пермиссивных микроусловий для дифференцировки и жизнеспособности остеобластов.
Др. исследования decorin- и lumican-дефицитных мышей подтвердили мнение, что SLRPs являются важными медиаторами клеточной пролиферации и жизнеспособности. Это согласуется с находками, продемонстрировавшими, что эктопическая экспрессия decorin фибробластах, линиях злокачественных клеток и опухолевых xenografts супрессирует рост (Ameye & Young 2002, Reed & Iozzo 2002), дефицит decorin индуцирует пролиферацию фибробластов в перидонтальных связках (Hakkinen et al. 2000) и ускоряет lymphoma туморогенез у мышей, лишенных гена опухолевого супрессора p53 (Iozzo et al. 1999). С др. стороны, отсутствие decorin при обструктивной нефропатии у животных моделей приводит к повышенному апоптозу в эпителиальных клетках канальцев, связанному с низкими уровнями p27
KIP1 и повышенной экспрессией TGF-β1, это прямо указывает на защитную роль decorin в fibrotic болезни почек посредством как TGF-β-зависимого, так и -независимого механизмов (Schaefer et al. 2002). В роговице, lumican регулирует пролиферацию и апоптоз стромальных кератоцитов во время заживления ран (Vij et al. 2004). Lumican-нулевые мыши обнаруживают повышенную пролиферацию этих клеток, вызванную подавлением белка p53, контролирующего клеточный цикл, и его нижестоящей мишени p21
WAF1/CIP1. Апоптоз также снижается в мутантной роговице из-за негативной регуляции пути Fas/Fas lлиганда. Lumican может влиять на этот путь или путем взаимодействия с Fas, модулируя тем самым количество Fas на клеточной поверхности, или путем связывания растворимого Fas лиганда и предоставления его Fas.
Microfibrils and Growth Factor Signaling
Всё увеличиваются генетические доказательства, что богатые fibrillin микрофибриллы играют важную роль в передаче сигналов TGF-β/BMP (rev. Judge & Dietz 2005, Kielty et al. 2002, Ramirez et al. 2004). TGF-βs секретируются в латентной форме в комплексе, который включает LTBP-1, -3, или -4. Эти латентные комплексы секвестрируются в ВКМ; после активации, TGF-β высвобождается и инициирует нижестоящий сигнальный каскад путем взаимодействия с рецепторами клеточной поверхности (Annes etal. 2003). Структурная гомология между fibrillins и LTBPs и способность LTBPs связывать fibrillins ведет к предположению, что микрофибриллы играют роль в передаче сигналов TGF-β. В самом деле, гипоморфные по fibrillin-1 мыши характеризуются Marfan-подобной эмфиземой и prolapse митрального клапана, ассоциированными с повышенной активацией TGF-β (Neptune et al. 2003, Ng et al. 2004). И легочный и сердечный фенотипы могут быть устранены применением нейтрализующих TGF-β антител, это подтверждает, что fibrillin-1-LTBP взаимодействия важны для секвестрации латентного комплекса и иммобилизации TGF-β. Ltbp-3-/- и гипоморфные Ltbp-4 мыши также обнаруживают эмфизему, которая, по-видимому, ассоциирует со снижением передачи сигналов TGF-β (Colarossi et al. 2005, Sterner-Kocketal. 2002). Интересно, что LTBP-4 дефицит специфически блокирует секрецию и активацию TGFβ-1, это сопровождается повышением количества BMP4 и усилением экспрессии генов мишеней для BMP4 (Koli et al. 2004). Эти находки указывают на то, что легочный фенотип частично обусловлен переключением с TGF-β на передачу сигналов BMP4. Всё это подтверждает, что fibrillin-1 и LTBPs надежно контролируют уровни факторов роста во время развития легких и что отклонения от обычной физиологической концентрации TGF-β могут иметь некоторые патологические последствия в зависимости от повышения или понижения концентрации.
Дополнительным подтверждением роли fibrillins/LTBPs в модуляции передачи сигналов ростового фактора получены при анализе
Fbn2 и Ltbp-3 нокаутных мышей.
Ltbp-3-нулевые мыши характеризуются костными аномалиями, включая черепно-лицевые уродства, остеоартриты и остеопетроз, как результат снижения передачи сигналов TGF-β (Dabovic et al. 2002,2005). Fibrillin-2-жефицитные мыши обнаруживают билатеральную синдактилию, ассоциированную с нарушенной дифференцировкой мезенхимы, возможно благодаря отсутствию функциональных взаимодействий между дизорганизованными fibrillin-2-дефицитными микрофибриллами и ростовыми факторами (Arteaga-Solis et al. 2001). Мыши, дважды гетерозиготные по
Fbn2 and Bmp-7, обнаруживают синдактилию и полидактилию (характерный фенотип для соотв. мутантов, гомозиготных по одному из генов, соотв.), демонстрируя тем самым, что нарушение передачи сигналов BMP-7 может быть ,по крайней мере частично, ответственным за синдактилию у
Fbn2-/- мышей (Arteaga-Solis et al. 2001).
Laminins in Signaling
Соединение laminin с integrin и nonintegrin рецепторами клеточной поверхности запускает множественные пути сигнальной трансдукции, модулирующие клеточное поведение как при физиологических, так и патологических процессах. Огромное количество данных in vitro указывает на участие laminin-индуцированных сигналов в дифференцировке, миграции и пролиферации клеток; в регуляции клеточной экспрессии; а также в прогрессировании опухолей и ангиогенезе (Ekblom et al. 2003, Givant-Horwitz et al. 2005). Менее известно по передаче сигналов laminin in vivo, но недавние эксперименты на мышах пролили свет на некоторые детали развития волос и периферических нервов.
Laminin-511 является основным ламинином в BM ниже удлиняющегося эпителия зачатка волос. Развивающаяся кожа у мышей, дефицитных по поздней эмбриональной летали α5 цепи, содержит достоверно меньше зачатков волос, чем контрольная кожа, а трансплантаты α5-null кожи не дают волос у мышей nude (Li et al. 2003).
Использование антител против laminin-511 человека приводит к сходному блокированию роста волос в трансплантатах кожи скальпа человека. Последующие исследования показали, что неспособность к элонгации зачатков волос α5-мутантных трансплантатов сопровождается потерей ассоциированной с волосяным фолликулом клеточной пролиферации и передачи сигналов sonic hedgehog (Shh) signaling (Li et al. 2003). Shh является критическим морфогеном для пролиферации во время развития волос; Shh-нулевые мыши обнаруживают нарушение элонгации зачатков (Chiang et al. 1999). Laminin α5-дефицитная эмбриональная кожа и трансплантаты не экспрессируют Shh или его ген мишень Gli1 (Li et al. 2003), демонстрируя, что laminin-511 необходим для Shh-управляемого морфогенеза волосяных фолликулов, возможно посредством соединения с интегринами, т.к. потеря α3 bkb β1 integrin ведет к сходным дефектам волосяных фолликулов (Brakebusch et al. 2000, Conti et al. 2003).
Многие доказательства указывают на то, что богатые laminin BMs, синтезируемые Шванновскими клетками, играют критическую роль в миэлинизации аксонов во время развития периферических нервов (Miner & Yurchenco 2004). Мыши и люди, лишенные laminin α2 цепи и мыши с мутацией, специфической для Шванновских клеток, в гене, кодирующем γl цепь, характеризуются dysmyelinating периферической нейропатией. Элиминация γl цепи в Шванновских клетках приводит к истощению всех изоформ ламинина, что конечно же нарушает взаимодействие между аксонами и Шванновскими клетками и вызывает арест Шванновских клеток на promyelinating стадии из-за неспособности подавлять способствующий дифференцировке транскрипционный фактор Oct-6 (Yu et al. 2005). Отсутствие собственно взаимодействий между аксонами и Шванновскими клетками снижает фосфорилирование β-neuregulin-1 рецепторов ErbBl и ErbB3, это ведет к снижению пролиферации Шванновских клеток. На постнатальных стадиях мутантные Шванновские клетки характеризуются нарушением передачи сигналов? управляемой PI3-kinase/Akt, это ведет к активации проапоптического caspase каскада. Т.к инъекции laminin пептидов в мутантный седалищный нерв частично устраняют апоптоз, то индуцированные ламинином сигналы обычно участвуют в регуляции жизнеспособности Шванновских клеток
Fibronectin
Fibronectins (Fns) являются крупными, адгезивными гликопротеинами, которые существуют в двух формах: в качестве растворимых плазменных Fn (pFn) в крови и нерастворимых клеточных Fn (cFn) во ВКМ. Fn влияет на клеточные активности и организацию матрикса путем взаимодействия с интегринами, syndecans и молекулами ВКМ. Мыши, лишенные всех изоформ Fn, погибают на E8.5 от тяжелых дефектов миграции клеток и формирования мезодермы (George et al. 1993), демонстрируя тем самым критическую роль Fns во время средины эмбриогенеза. Интересно, что условно нокаутные мыши, лишенные pFn (Sakai et al. 2001) или cFn в хондроцитах (Aszodi et al. 2003) выглядят нормальными, указывая, что эти изоформы, по-видимому, несущественны для гемостаза и развития хряща, соотв. Однако, pFn вносит вклад в некоторые репаративные процессы. У модельных мышей повреждения артерий, pFn дефицит снижают рост и стабильность тромбов при касательных напряжениях из-за нарушенной pFn-обуславливаемой слипчвивости среди тромбоцитов (Ni et al. 2003). Вследствие церебральной ишемии отсутствие pFn увеличивает размеры инсульта, это ассоциирует с повышенным уровнем апоптоза как нервных, так и не-нейрональных клеток (Sakai et al. 2 001 }. Потеря взаимодействия между pFn и α5β1 integrin снижает экспрессию антиапоптическго белка Bcl-2 и приводит к активации эффектора апоптоза caspase-3. Т.о., pFn поддерживает клеточную жизнеспособность в местах повреждения головного мозга посредством integrin-опосредованных сигналов.
COLLAGEN-DERIVED MATRICRYPTINS AND ANGIOGENESIS
В последнее время получены доказательства, что отдельные домены внутри ВКМ белков могут передавать сигналы посредством рецепторов клеточной поверхности и регулировать биологические функции. Эти т.наз. matrikine субдомены или действуют непосредственно с нативных ВКМ макромолекул (natural matrikines) или д. сначала подвергнуться конформационным изменениям или протеолитическому процессингу (cryptic matrikines) (Tran et al. 2005).
В последнюю декаду большая часть внимания была сконцентрирована на скрытых фрагментах, генерируемых с NC1 домена collagen IV αl/α2/α3 цепей (называемых arresten, canstatin и tumstatin, соотв.), collagen XV (restin) и collagen XVIII (endostatin) (Figure 3а) (Iozzo 2005). Эти циркулирующие в крови фрагменты ингибируют ангиогенез in vitro и in vivo и редуцируют опухолевый рост у животных моделей (Marneros & Olsen 2001). Неожиданно мыши, лишенные α3 цепи collagen IV (Hamano et al. 2003), collagen XV (Eklund et al. 2001), collagen XVIII (Fukai et al. 2002), или в обих коллагенах XV и XVIII (Ylikarppa et al. 2003) не обнаруживают очевидных дефектов в физиологическом ангиогенезе, подтверждая, что расщепленные NC1 фрагменты не являются существенными компонентами путей, регулирующих ангиогенез. Тем не менее более детальный анализ выявил, что их функция может быть ограничена определенными тканями и/или имеет отношение к определенным патологическим состояниям.
Мыши, лишенные tumstatin-продуцирующей α3 цепи коллагена IV, обнаруживают усиленные опухолевый рост и ассоциированный с опухолями ангиогенез по сравнению с мышами дикого типа (Hamano et al. 2003). Применение физиологических концентраций tumstatin к нулевым мышам снижает как количество кровеносных сосудов, так и скорость роста опухолей до уровней дикого типа, это подтверждает формально, что tumstatin домен сам по себе является эндогенным супрессором патологического ангиогенеза. Tumstatin отщепляется от α3(IV) цепи с помощью BM-деградирующих MMPs (Hamano et al. 2003). MMP-9-дефицитные мыши обнаруживают пониженные уровни циркулирующих в крови tumstatin levels и обнаруживают ускоренный рост опухолей, который может быть возвращен к уровню дикого типа путем увеличения tumstatin до нормальной физиологической концентрации с помощью экзогенного введения (Hamano et al. 2003). Т.о., MMPs, которые в целом. как полагают, являются позитивными регуляторами опухолевого ангиогенеза, могут выполнять противоположную функцию путем высвобождения эндогенных антиангиогенных фрагментов, таких как tumstatin (Hamano & Kalluri 2005).
Col18a1-нулевые мыши характеризуются дефектами в развитии васкулатуры задних частей глаз (rev. Marneros, Olsen 2005), указывая на роль коллагена XVIII/endostatin в модуляции васкуляризации специфических анатомических мест. Аномалии проявляются постнатально и включают задержку регрессии hyaloid сосудов вдоль поверхности сетчатки, а также нарушенный рост и паттерн сосудистой сети сетчатки. Интересно, что дефекты, сопровождаемые пониженной экспрессией VEGF в нейроглие (Fukai et al. 2002), которая обычно позитивно регулируется с помощью гипоксии в начале регрессии hyaloid сосудов и стимулирует сосудистые разрастания от головки зрительного нерва. Механизм, лежащий в основе сосудистой регрессии у Col18al-нулевых мышей, неясен, но эксперименты с эксплантами аорты и легочных эндотелиальных клеток, выделенных от мышей дикого типа и мутантных мышей (Li & Olsen 2004) дают некоторые указания. Эта модель характеризуется динамическим процессом элонгации и регрессии микрососудов в аортальных эксплантах в условиях среды, лишенной сыворотки. Мутантные экспланты обнаруживают усиленное разрастание сосудов, которое устраняется добавлением физиологических уровней endostatin в культуру. Более того, изолированные легочные эндотелиальные клетки от Col18al-нулевых мышей обнаруживают повышенную адгезию к fibronectin по сравнению с клетками дикого типа. Т.к. пролиферация и миграция мутантных эндотелиальных клеток нормальная, то в соответствии с этими находками повышенная слипчивость эндотелиальных клеток к FN-содержащему матриксу стабилизирует вновь формируемые сосуды и снижает регрессию, обусловливая общее увеличение разрастаний микрососудов. Т.о., collagen XVII/endostatin могут осуществлять антиангиогенный эффект, модулируя взаимодействия между энгдотелиальными клетками и подлежащим ВКМ из сосудистых basement мембран.
In vitro исследования показали, что у людей endostatin и arresten взаимодействуют с α5β1 и αlβ1 integrins, соотв. и ингибирует миграцию EC, блокируя FAK/Ras/Raf/ERKl/p38 митогеном активруемый протеин киназный путь (Sudhakar et al. 2005. Sudhakar et al. 2003). Endostatin индуцирует также разборку актинового цитоскелета путем подавления активности RhoA activity (Wickstrom et al. 2003). Tumstatin взаимодействует с αvβ3 integrin, приводя к ингибированию FAK/PI3K/Akt/mTOR-обусловленного пути синтеза белков (Figure 3b) (Alaeshima et al. 2002, Sudhakar et al. 2003). В согласии с
in vitro находками, endostatin, но не tumstatin ингибирует неоваскуляризацию в Matrigel plugs, имплантированных β3 integrin-дефицитным мышам (Hamano et al. 2003), a arresten неспособен ингибировать ангиогенез и опухолевый рост у мышей, лишенных αl integrin субъединицы (Sudhakar et al. 2005). Следовательно, имеются хорошее доказательство, что биоактивные NC1 фрагменты осуществляют свой антиангиогенные эффекты посредством модуляции integrin-зависимых сигнальных путей. Более того, недавние исследования показали, что антиангиогенная функция этих субстанций может зависеть адгезивных белков плазмы. Напр., endostatin и anastellin (антиангиогенный фрагмент, происходящий из fibronectin) не ингибируют васкуляризацию Matrigel plug у мышей, лишенных pFn (Yi et al. 2003). В предлагаемой модели pFn служит в качестве кофактора, который поставляет ангиостатические фрагменты в места ангиогенеза и презентирует их интегириновым рецепторам (Akerman et al. 2005).
MATRICELLULAR PROTEINS
Термин matricellular белки приложим к определенным членам некоторых семейств ВКМ белков, которые прежде всего модулируют клеточные функции и не играют определенно структурной роли в матриксе. Разные группы matricellular белков сегодня включают thrombospondin (TSP) 1 и 2, SPARC (известный также как BM-40), SCl/hevin, tenascin C и X, osteopontin, и члены семейства белков CCN (cyr61, ctgf, nov). Matricellular белки обладают некоторыми общими признаками. Напр., (a) они обнаруживают высокие уровни экспрессии во время эмбриогенеза, которя постепенно снижается постнатально; (b) они соединяются с рецепторами клеточной поверхности, ростовыми факторами, цитокинами и протеазами; (c) они обеспечивают de-adhesion клеток, зависящих от прикрепления; и (d) их элиминация из матрикса обычно несовместима с жизнью (Bornstein & Sage 2002, Bornstein et al. 2004).
TSPl и TSP2 являются гетеротримерными молекулами, формирующими разные подгруппы внутри семейства thrombospondin. И TSPl и TSP2 являются мощными ингибиторами ангиогенеза in vitro, a у мышей с их дефицитом увеличивается плотность сосудов в коже [TSPl (Crawford et al. 1998)] или во множественных тканях [TSP2 (Kyriakides et al. 1998)]. В глазах TSPl и TSP2 управляют индуцированным воспалени ем роговицы и онтогенетическим ангиогенезом радужки, соотв. (Cursiefen et al. 2004). TSPl- и TSP2-нулевые мыши характеризуются повышенным туморогенезом (Hawighorst et al. 2001, Rodriguez-Manzaneque et al. 2001), тогда как избыточная экспрессия TSPl у мышей склонных к опухолям молочных желез снижает показатель опухолей (Rodriguez-Manzaneque et al. 2001). Более того, TSP1/TSP2 обладают самостоятельными ролями во время заживления ран: отсутствие TSPl или обоих TSPl и TSP2 нарушает раннюю фазу реакции на ранение, это приводит к задержке заживления, тогда как TSP2-нулевые раны обнаруживают повышенную неоваскуляризацию и ускоренное заживление (Agah et al. 2002). В целом эти данные показывают, что TSPl и TSP2 в самом деле являются ангиогенными регуляторами in vivo.
Фенотипы др. matricellular нокаутных мышей обнаруживают сходства. Усиленный опухолевый рост и ускоренное закрытие ран описаны у SPARC-нулевых мышей (Bradshaw et al. 2002, Brekken et al. 2003), a tenascin-X нокауты обнаруживают повышенную инвазию опухолей (Matsumoto et al. 2001). Tenascin-X дефицит вызывает снижение содержания collagen I в коже, приводящее к сверхрастяжимости кожи и воспроизведению нарушения при мутации гена TNXB, вызывающего синдром Ehlers-Danlos (Mao et al. 2002). Уменьшение collagen I описано в дермисе и жировых подушках SPARC-нулевых мышей, это приводит нарушению предела прочности на разрыв кожи и ожирения, соотв. (Bradshaw et al. 2003). TSP2 нокаутные мыши также обладают ломкой кожей, что связано с дизорганизованной архитектурой кожного коллагена (Kyriakides et al. 1998). Кроме того, многие matricellular белки экспрессируются в скелете и соотв. нокаутные мыши характеризуются скелетными аномалиями во время развития и регенерации кости (Alford & Hankenson 2006).
Хотя механистические детели, лежащие в основе дефектов, неясны, несколько линий доказательств указывают на то, что matricellular белки осуществляют свои биологический функции путем регуляции активности ростовых факторов и протеаз, которые в свою очередь влияют на пролиферацию/выживаемость клеток и отложение ВКМ. Напр., TSPl соединяется и активирует латентный activates latent TGF-βl in vitro (Annes et al. 2003, Hyytiainen et al. 2004). In vivo, TSP1- and TGF-β1-нулевые мыши обладают перекрывающимся фенотипом (напр., в легких и поджелудочной железе), a дефекты у нокаутных TSP1 мышей могут быть частично устранены с помощью TGF-β1-активирующего пептида, происходящего из TSP1, это подтверждает мнение, что TSP1 in vivo является латентным активатором TGF-β1 (Crawford et al. 1998). Напротив, SPARC взаимодействуют с TGF-β1-рецепторным комплексом в присутствии TGF-β1, который в свою очередь модулирует активацию нижестоящего сигнального каскада (Francki et al. 2004). Растут доказательства. указывающие, что matricellular белки могут контролировать уровни экспрессии MMPs (Bomstein et al. 2004). TSP2-нулевые ткани обнаруживают повышенные уровни MMP-2, что ведет к MMP-2-индуцированному протеолизу тканевой transglutaminase, энзима. участвующего в стабилизации ВКМ, это предоставляет пригодное объяснение аномалий матрикса. наблюдаемых в TSP2-дефицитной коже (Agahet al. 2005). Сходным образом, уровни MMP-2 (и MMP-9) были повышены в злокачественных глиомах, накапливающихся у TSP2-нулевых мышей (Fears et al. 2005). Согласно существующей модели TSP2 контролирует уровни MMP-2 обеспечивая обусловленный lipoprotein receptor-related protein (LRP) низкой плотности эндоцитоз MMP-2. Напротив, TSP1 супрессирует процессинг pro-MMP-9 с помощью MMP-3; Следовательно, в отсутствие TSP1 увеличиваются уровни активной MMP-9 во время туморогенеза, облегчается как ангиогенез, так инвазия опухолей (Rodriguez-Alanzaneque et al. 2001). Усиление метастазирования у tenascin-X-нулевых мышей связано с повышением активности MMP-2 и MMP-9 (Matsumoto et al. 2001).
Роль matricellular белков в ЦНС недавно идентифицирована благодаря экспериментам с нокаутами. TSP1/TSP2 секретируются астроцитами и способствуют синаптогенезу in vivo и in vitro (Christopherson et al. 2005). Эти две молекулы присутствуют в ЦНС мышей, когда устанавливаются синапсы, а в головном мозге TSP1/TSP2 двойных нулевых мышей формируется приблизительно на 40% меньше синапсов, чем в головном мозге мышей дикого типа. TSPs, по-видимому, действуют в сочетании с др. сигналами, происходящими из астроцитов, чтобы продуцировать функциональные синапсы в ЦНС (Christopherson et al. 2005). Мыши, лишенные SCl/hevin не имеют обнаружимого фенотипа. Церебральный кортекс таких животных, однако, обнаруживает неправильное смещение нейронов, указывающее на то, что SCI dfяважным миграции нейронов в развивающейся коре благодаря своей антиадгезивной активности (Gongidi et al. 2004). Наконец, устранение tenascin-C или tenascin-R (который также может быть matricellular белком) ведет к различным нейрональным дисфункциям (Evers et al. 2002, Saghatelyan et al. 2004).
Среди matricellular белков два члена семейства CCN, CCX1 и CCN2, по-видимому, существенны для жизни. Разрушение CCN 1 (CYR61) вызывает пренатальную летальность благодаря нарушениям сосудистого развития плаценты и эмбрионов (Mo et al. 2002). CCN2 [или CTGF (connective tissue growth factor)]-нулевые мыши погибают при рождении и имеют скелетные аномалии, такие как аномальное ремоделирование матрикса и ангиогенеза в ростовых пластинках (Ivkovic et al. 2003). Высокая доза апоптоза сосудистых клеток у Ccnl
-/-мышей и пониженная скорость пролиферации Ccn2
-/- хондроцитов показывают, что общая функция CCN белков состоит в контроле клеточной гибели и выживаемости зависимым от контекста образом. В самом деле, эксперименты с клеточными культурами показали, что CCN1 индуцирует апоптоз в фибробластах, в то время как он способствует выживанию эндотелиальных клеток
(Todorovic et al. 2005).
CONCLUDING REMARKS
The molecular constituents of the ECM form larger complexes that provide structural and instructive signals for tissue differentiation, development, and function. The composition of the ECM and the distribution of its components define tissue architecture and cellular responses for cytokine/growth factor stimulation. The complexity of the ECM makes it difficult to identify the functional repertoire of ECxM proteins by biochemical or cell culture experiments. Hence, mouse models are particularly useful for analyzing the biological significance of ECM components in a wide variety of in vivo processes. Mouse strains with engineered mutations in genes encoding ECM proteins have shown that most ECM components have roles beyond a simple structural scaffold and have sometimes revealed unexpected functions that cannot be predicted by in vitro approaches. Knockout mice and cell lines established from them have helped to elucidate intracellular signal transduction cascades activated by ECM-cell interactions, In the future, the analysis of mice earning null mutations in ECM molecules, in combination with knock-in approaches to address specific mutations, will enable us to identify the mechanisms driving a wide variety of pathologies. The involvement of ECM in regulating and maintaining tissue homeostasis will suggest novel therapies with which to combat diseases with an abnormal ECM component.
SUMMARY POINTS
1. Extracellular matrix proteins are modular, multifunctional molecules that provide a structural as well as an instructive environment for cells and tissues.
2. Genetically modified mice provide an excellent in vivo approach with which to discover the functions of ECM proteins for developmental, physiological, and pathological processes.
3. The mechanical stability of connective tissues and basement membranes is determined by the proper assembly of ECM proteins into interactive macromolecular architectures.
4. ECM components integrate growth factor- and integrin-mediated signaling pathways by controlling the presentation of growth factors to cells as well as by binding directly to cell surface receptors such as integrins.
5. The antiangiogenic role of collagen-derived matrikines is context dependent and involves integrin-mediated signaling cascades.
6. Matricellular proteins modulate cell functions primarily by regulating growth factor and MMP activities.
Сайт создан в системе
uCoz 