Большая часть скелета млекопитающих формируется благодаря процессу эндохондральной оссификации. Будущие кости закладываются в качестве хрящевой матрицы, а клетки внутри этих предшественников-хондроциты-проходят через жестко контролируемые фазы пролиферации и дифференцировки, которые регулируют продольный рост эндохондральных костей (Kronenberg, 2003; Beier, 2005; Provot and Schipani, 2005; Mackie et al.; 2008). В длинных костях конечностей (бедро, плечо и т.д.), самые наружные клетки (ближайшие к концам кости) пролиферируют с низкой скоростью и называются покоящимися хондроцитами (Fig. 1). Затем эти клетки увеличивают скорость своей пролиферации и делятся быстро вдоль продольной оси кости и формируют характерные столбцы клеток. В конечном итоге клетки выходят из клеточного цикла и вступают в терминальную дифференцировку, которая характеризуется изменениями генной экспрессии и драматическим увеличением клеточного объема, что позволило назвать их гипертрофическими хондроцитами. Гипертрофические хондроциты минерализуют свой внеклеточный матрикс (extracellular matrix (ECM)) и подвергаются апоптозу, в то время как область гипертрофического хряща наполняется кровеносными сосудами вместе с остеокластами (кость- и хрящ-резорбирующими клетками) и клетками предшественниками остеобластов (кость-формирующих клеток). Все вместе эти клетки деградируют и ремоделируют хрящевой ECM, а остеобластные клетки прилипают к остаткам хрящевого ECM, чтобы сформировать костную ткань в этом первичном центре оссификации.
Пролиферация и гипертрофия хондроцитов вместе с синтезом ECM являются основными силами, которые управляют ростом эндохондральной кости (Hunziker, 1994; Ballock, 2003; van der Eerden, 2003). После образования вторичных центров оссификации на концах длинных костей, происходят события жизненного цикла хондроцитов, описанные выше (пролиферация, гипертрофия и апоптоз), пространственно организованным способом в структуре, называемой ростовой пластинкой, расположенной между первичным и вторичным центром оссификации (Fig. 2). Активность ростовой пластинки, т.о., контролирует продольный рост кости и в конечном итоге высоту взрослого, а многочисленные нарушения, характеризуемые задержкой роста и уменьшенной финальной высотой, имеют в своей основе нарушения физиологии хондроцитов или обнаруживают патологические изменения ростовой пластинки, вторичные по отношению к др. причинам. Поэтому наиболее необходимо понять механизмы, контролирующие пролиферацию и дифференцировку хондроцитов, чтобы достичь наилучшего ведения этих болезней (Ballock, 2003; van der Eerden, 2003; Nilssonetal., 2005).
Помимо своего вклад в рост костей, гипертрофические хондроциты координируют множественные аспекты эндохондральной оссификации посредством секретируемых ими продуктов (Chung, 2004). ECM, откладываемый гипертрофическими хондроцитами служит в качестве матрицы для последующего образования кости и эти клетки также секретируют многие растворимые белки, включая vascular endothelial growth factor (VEGF), Indian hedgehog и лиганд receptor activator of NF-kB (RANK) , который контролирует активности др. клеточных клонов, участвующих в эндохондральной оссификации (напр..эндотелиальные клетки, остеобласты и остеокласты, соотв. ). Механизмы, которые регулируют переход к гипертрофии и экспрессии генов. специфичных для гипертрофических хондроцитов, являются поэтому областью активных исследований многих лабораторий.
В последнюю декаду идентифицированы две группы транскрипционных факторов, которые контролируют ключевые ступени дифференцировки хондроцитов. Ген
Sox9 абсолютно необходим для формирования хряща и способствует ранним стадиям дифференцировки хондроцитов вместе с родственными
Sox5 и Sox6 генами. Runx2 и родственный ему Runx3 способствуют дифференцировке гипертрофических хондроцитов, помимо их функции в др. тканях (наиболее известна регуляция остеогенеза с помощью Runx2). Опубликовано несколько обзоров по этим транскрипционным факторам (Yang and Kar-senty, 2002; Lefebvre and Smits, 2005; Schroeder et al., 2005; Yoshida and Komori, 2005; Komori, 2006). Кроме того, многочисленные хорошо изученные транскрипционные факторы, которые обеспечивают клеточные реакции на внеклеточные сигналы, были продемонстрированы и постулирована их роль в дифференцировке хондроцитов. Сюда входят белки Smad (передача сигналов TGFβ / BMP) и Gli (hedgehog) семейство, β -catenin и взаимодействующие с ним факторы, такие как LEFs/TCFs (Wnt) и hypoxia-inducible factor 2a (Hiflα ) в качестве медиатора реакции хондроцитов на гипоксию и некоторые ядерные рецепторы (напр., рецепторы тироидных гормонов, витамин D, глюкокортикоиды, ретиноевая кислота и эстроген) (Beier et al., 1999a; Juul, 2001; Robson et al., 2002; Yang and Karsenty, 2002; Ballock, 2003; van der Eerden, 2003; Kobayashi and Kronenberg, 2005; Lefebvre and Smits, 2005; Schipani, 2005). Наши усилия направлены на то, чтобы понять транскрипционные сети, управляющие дифференцировкой хондроцитов и эндохондральной оссификацией. Необходимо отметить, понимание механизмов, которые
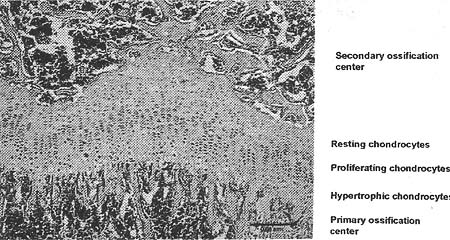 Figure 2. The growth plate. Hematoxylin and Eosin staining of a paraffin section from the humerus of a 10-day old mouse shows the primary and secondary ossification centers and the zones of resting, proliferating, and hypertrophic chondrocytes.
Figure 2. The growth plate. Hematoxylin and Eosin staining of a paraffin section from the humerus of a 10-day old mouse shows the primary and secondary ossification centers and the zones of resting, proliferating, and hypertrophic chondrocytes.
контролируют гипертрофию хондроцитов, не только важны в отношении развития и роста скелета, но и также имеет большое значение для дегенерации хряща во время патогенеза остеоартритов (Drissi et al., 2005). Это подчеркивается тем фактом, что мыши, у которых одна копия гена
Runx2 была инактивирована, частично защищены от деградации хряща в хирургических моделях остеоартритов (Kamekura et al., 2006). Т.о., др. транскрипционные факторы, которые способствуют гипертрофии хондроцитов, также могут участвовать в прогрессировании остеоартритов и поэтому представляют собой новые мишени для разработки болезнь-модифицирующих лекарств.
SHOX AND SHOX2
Мутации потери функции гена SHOX (short stature homeo-box) у людей были идентифицированы как причина нескоторых болезней, характеризующихся низким ростом и скелетными деформациями (rev. Marchinietal., 2007). Сюда входят и низкий рост, наблюдаемый у пациентов с синдромом Turner, Leri-Weill dyschondrosteosis, Langer mesomelic dysplasia, и в некоторых случаях идиопатического низкого роста (Rao et al., 1997; Belin et al., 1998; Shears et al., 1998; Stuppia et al., 1999; Clement-Jones et al., 2000; Rap-pold et al., 2002; Zinn et al., 2002; Munns et al., 2003). Мутации, вызывающие болезнь у людей включают крупные делеции, также как и nonsense и missense мутации в кодирующей области. Важно, что гаплонедостаточность SHOX уже вызывает задержку роста, демонстрируя важную регуляторную функцию активности SHOX в нормальной эндохондральной оссификации.
Мышиный геном не содержит гена Shox, но содержит его близкого родственника, Shox2. Две группы недавно описали скелетные дефекты у мышей при специфической для конечностей или повсеместной инактивации гена Shox2 (Cobb et al., 2006; Yu et al., 2007). В обоих случаях недостаточность Shox2 приводила к драматическому укорочению костей проксимальных частей конечностей (напр., плечевых и бедренных костей), в то время как дистальные части костей повреждены в значительно меньшей степени. На клеточном уровне гипертрофия хондроцитов сильно снижена у мутантов.,
Уменьшенная гипертрофия скорее всего обусловлена более низкими уровнями Runx2 и Runx3. Интересно, что паттерн затрагиваемых костей отличен от такового у людей с мутациями SHOX (который распространяется на более дистальные части костей), и скорее всего обусловлен тем фактом, что ген Shox2 мышей обнаруживает др. паттерн экспрессии, чем ген SHOX у людей.
Missense мутации у пациентов, как было установлено, влияют на ядерную локализацию, димеризацию, связывание ДНК и активацию транскрипции с помощью SHOX (Sabherwal et al., 2004; Schneider et al., 2005). На клеточном уровне избыточная экспрессия белка SHOX вызывает арест клеточного цикла и апоптоз в ряде клеточных типов, включая хондроциты (Marchini et al., 2004). Эта активность согласуется с позитивной регуляцией экспрессии
Shox в гипертрофических хондроцитах и с фенотипом пониженной гипертрофии у Shox2-нулевых мышей. Эти данные указывают на то, что белки Shox способствуют выходу из клеточного цикла и гипертрофической дифференцировке. Как подчеркивалось ранее, это по-видимому, происходит, по крайней мере, частично, путем стимуляции экспрессии
Runx2 и 3, также как и определенных ингибиторов клеточного цикла (Marchini et ai., 2004; Cobb et al., 2006; Yu et al., 2007). Однако неясно, являются ли эти гены непосредственными генами мишенями для белков Shox. Более того, оба паттерна экспрессии
Shox/Shox2 и спектр затрагиваемых костей при Shox недостаточности указывает на то, что эти белки не контролируют экспрессию
Runx2/3 во всех эндохондральных костях. Т.о., хотя и продемонстрирована важность SHOX для эндохондральной оссификации и роста костей и получены некоторые указания на его клеточную функцию, однако молекулярные механизмы действия выше и ниже SHOX и SHOX2 ещё предстоит выяснить.
DLX5 AND DLX6
Подобно Shox/Shox2, distal-less гомеобоксные белки, Dlx5 и 6, являются гомеодоменовыми транскрипционными факторами с различными ролями в конечностях и в развитии скелета. Одновременная инактивация обоиз из этих генов ведет к тяжелым дефектам скелета и конечностей (Robledo et al., 2002), но неясно до какой степени эти дефекты обусловлены скорее измененным формированием паттерна тела, чем прямыми эффектами на клеточную дифференцировку. Недавно было показано, что избыточная экспрессия Dlx5 в хрящах кур и мышей способствует гипертрофии хондроцитов, тогда как недостаточность Dlx5 ведет к задержке гипертрофии (Ferrari and Kosher 2002; Bendall et al., 2003; Chin et al., 2007). Исследования in vitro покзали, что Dlx6 обладает сходной с Dlx5 способностью стимуляции дифференцировки хондроцитов (Hsu et al., 2006), а in vivo it он, по-видимому, частично компенсирует потерю Dlx5 в остеобластах (Li et al., 2008), но неясно выполняет ли эндогенный ген Dlx6 функцию в развитии хрящей.
Исследования остеобластов указывают на то, что Dlx5 индуцирует экспрессию
Runx2 также как и второго остеогенного транскрипционного фактора, Osterix, скорее всего посредством непосредственного соединения с соотв. промоторами (Lee et al., 2003, 2005; Ulsamer et al., 2008). Кроме того, Runx2 и Dlx5 белки, по-видимому, взаимодействуют непосредственно, чтобы контролировать транскрипцию гена, кодирующего bone sialoprotein (BSP), маркер остеобластов и гипертрофических хондроцитов (Roca et al., 2005). Не было исследований, адресованных потенциальному синергичному, комплементарному или аддитивному действию белков Shox и DIx, учитывая, что оба семейства являются гомеодоменовыми факторами.
NKX3.2/BAPX1
В противоположность генам SHOX и DIx др. гомеобелок, Nkx3.2 (известный также как Bapx1), экспрессируется прежде всего в пролиферирующих хондроцитах длинных костей конечностей (Provot et al., 2006). Nkx3.2 по-видимому, обеспечивает передачу сигналов от паракринного фактора роста PTHrP (parathyroid hormone-related peptide) , что предупреждает хондроциты от выхода из клеточного цикла и инициации дифференцировки. Избыточная экспрессия Nkx3.2 ведет к снижению экспрессии Runx2, а избыточная экспрессия Runx2 может противодействовать эффектам избыточной экспрессии Nkx3.2 в созревающих хондроцитах (Provot et al., 2006). Эти данные указывают на то, что репрессия Runx2 необходима для эффектов Nkx3.2 на хондроциты. Методы сдвига электрофоретической подвижности и иммунопреципитации хроматина указывают на то, что Nkx3.2 непосредственно соединяется с промотором Runx2, указывая тем самым, что репрессия Runx2 скорее всего является непосредственной (Lengner et al., 2005).
Недавнее исследование продемонстрировало др. роль Nkx3.2 в хондроцитах за счет непосредственного связывания субъединицы nuclear factor-kB (NFkB) , RelA (p65) и активации её независимым от лиганда способом (Park et al., 2007). Это взаимодействие ингибирует апоптоз хондроцитов, но RelA, как было показано, способствует пролиферации и дифференцировке хондроцитов (Wu et al., 2007), тем самым возникает возможность, что взаимодействия Nkx3.2/RelA контролируют дополнительные аспекты физиологии хондроцитов помимо супрессии апоптоза.
MEF2C
Транскрипционные факторы семейства myocyte enhancer factor 2 (MEF2) , как было показано первоначально, контролируют развитие кардиальной и скелетных мышц, но недавно установлено их участие во многих др. онтогенетических процессах (McKinsey et al., 2002; Potthoff and Olson, 2007). Нарушение функции одного из членов семейства, MEF2C, в хрящах приводит к задержке гипертрофической дифференцировки в эндохондральных костях мышей, в то время как эктопическая активация этого белка вызывает преждевременную гипертрофию (Arnold et al., 2007). Этот фенотип является более тяжелым у мышей, которые также дефицитны по гену Mef2d . Потеря активности MEF2 в хондроцитах ведет к снижению экспрессии Runx2, помещая MEF2C и MEF2D иерархи чески выше Runx2 в развитии хряща (Arnold et al., 2007). Эти in vivo исследования подтверждаются более ранними in vitro данными нашей лаборатории, где мы продемонстрировали позитивную регуляцию Mef2c на уровне транскрипции во время дифференцировки хондроцитов и потребность в активности MEF2 для нормальной активности промотора collagen X (Stanton et al., 2004; James et al., 2005).
Интересно, что потеря активности MEF2C дает противоположный фенотип по сравнению с инактивацией гена Hdac4, который кодирует гистоновую деацетилазу, участвующую в транскрипционной репрессии благодаря модификации хроматина (Vega et al., 2004). Ещё более интересным является тот факт, что компаундные мутанты по обоим генам обнаруживают нормализованные фенотипы каждого из индивидуальных мутантов (Arnold et al., 2007), демонстрируя взаимодействие генов во время развития хряща. Это согласуется с биохим. исследованиями, показавшими, что HDACs регулирует активность MEF2 в миобластах (Lu et al., 2000a,b; Zhang et al., 2002).
Исследования черепно-лицевого развития показали, что MEF2C необходим для нормальной экспрессии Dlx5 и родственного Dlx6 в бранхиальных дугах (Verzi et al., 2007). Более того, MEF2C, Dlx5 и Dlx6, по-видимому, контролируют лицевое развитие кооперативным образом. Поскольку MEF2C и Dlx5, по-видимому, необходимы для гипертрофии хондроцитов, то было бы интересно проверить, не существуют ли сходные взаимоотношения и в хрящах.
THE ATF/CREB FAMILY
Гипертрофическая дифференцировка нуждается в выходе из клеточного цикла и прекращении пролиферации хондроцитов. Член семейства activating transcription factor (ATF)/cyclic AMP response element binding (CREB) белок, как было установлено существенен для нормальной пролиферации хондроцитов. В частности, ATF2 и CREB способствуют ходу клеточного цикла путем активации промоторов cyclin D1 и cyclin A (Beier et al., 1999b, 2000, 2001; Beier and LuValle, 2002; Ionescu et al., 2001, 2003). Более того, ингибирование функции ATF2 или CREB ведет к снижению пролиферации хондроцитов
in vivo (Reimold et al., 1996; Long et al., 2001). Недавние исследования микромассивов в нашей лаб. идентифицировали экспрессию дополнительных членов семейства ATF/ CREB в хондроцитах. В частности, ATF3 позитивно регулируется во время гипертрофической дифференцировки
in vitro и in vivo и супрессирует промоторы cyclin D1 и cyclin A, противодействуя тем самым активности ATF2/CREB (James et al., 2006). Эти данные указывают на то, что ATF3-и потенциально др. чены семейства-способствуют выходу из клеточного цикла и тем самым началу гипертрофической дифференцировки. Однако оценка
in vivo роли ATF3 в развитии хряща ещё предстоит.
API AND MAF PROTEINS
Fos и Jun белки формируют activator protein 1 (API) комплекс и уже давно известно, что контролируют развитие и функцию скелета (Wagner, 2002; Wagner and Eferl, 2005). В недавнем исследовании было показано, что делеция гена, кодирующего Fos белок Fra2 (Fosl2) у мышей, приводит к снижению гипертрофии хондроцитов, вызывая кифоз, карликовость и преждевременную гибель (Karreth et al., 2004). В противоположность некоторым др. транскрипционным факторам, описанным здесь (напр., MEF2C и SHOX), снижение гипертрофии у Fosl2 KO мышей не сопровождается изменениями экспрессии Runx2. Однако пост-транскрипционные эффекты активности Runx2 не могут быть исключены в этом смысле, в частности, т.к. белки API, как было показано, непосредственно взаимодействуют с Runx факторами (Hess et al., 2001). Напротив, Fra2 может контролировать гипертрофическую дифференцировку Runx-независимым способом.
В противоположность Fra2 исследования
in vitro показали. что Jun белки также как c-Fos супрессируют дифференцировку хондроцитов (Kameda et al., 1997; Thomas et al., 2000), а мыши с мутациями потери функции генов
c-Fos или JunB обнаруживают пониженную пролиферацию хондроцитов (Wang et al., 1992; Hess et al., 2003). Т.о., различные члены
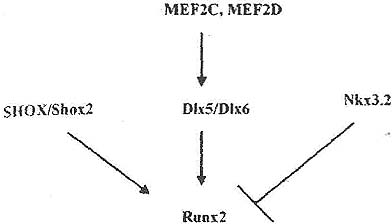 Figure 3. Runx2 expression is controlled by numerous upstream transcription factors. Studies in mice suggest that most, but not all, mutants that show altered hypertrophic chondrocyte differentiation display changes in Runx2 expression. For example, Runx2 expression is reduced upon inactivation of Shox2, Dlx5, and Mef2c genes as well as in response to overexpression of Nkx3.2. Although some of these transcription factors appear to bind directly to the Runx2 promoter (e.g., Dlx5 and Nkx3.2), this has not been shown for others. In addition, genetic or biochemical interactions between the different transcription factors discussed have not been examined in detail, although studies on branchial arch development suggest that MEF2C could act upstream of Dlx5/Dlx6.
Figure 3. Runx2 expression is controlled by numerous upstream transcription factors. Studies in mice suggest that most, but not all, mutants that show altered hypertrophic chondrocyte differentiation display changes in Runx2 expression. For example, Runx2 expression is reduced upon inactivation of Shox2, Dlx5, and Mef2c genes as well as in response to overexpression of Nkx3.2. Although some of these transcription factors appear to bind directly to the Runx2 promoter (e.g., Dlx5 and Nkx3.2), this has not been shown for others. In addition, genetic or biochemical interactions between the different transcription factors discussed have not been examined in detail, although studies on branchial arch development suggest that MEF2C could act upstream of Dlx5/Dlx6.
семейства API, по-видимому, играют контрастные роли в пролиферации и дифференцировке хондроцитов.
c-Maf и MafB являются родственными API транскрипционными факторами, которые, как было показано, позитивно регулируются во время дифференцировки хондроцитов (Sakai et al., 1997). Одна из изоформ c-Maf, как было показано, взаимодействует с Sox9, чтобы стимулировать транскрипцию collagen II (Huang et al., 2002), подтверждая тем самым роль c-Maf в ранней дифференцировке хондроцитов. Однако доминантный хрящевой фенотип c-Maf-дефицитных мышей характеризуется нарушенной гипертрофической дифференцировкой в соответствии с его паттерном экспрессии (MacLean et al., 2003). Эти расхождения мь. обусловлены специфическими функциями и биохимическими активностями разных изоформ c-Maf, необходимы дальнейшие исследования для решения этого вопроса. Кроме того, гетеродимерный партнер Maf белков, транскрипционный фактор nuclear factor E2 p45-related factor 2 (Nrf2), обнаруживает пониженную экспрессию во время дифференцировки хондроцитов и ингибирует различные аспекты дифференцировки хондроуитов
in vitro (Hinoi et al., 2007). Необходимо отметить экспрессию в дегенерирующем хряще (Appleton et al., 2007b), которая согласуется с нашими наблюдениями гипертрофической дифференцировки в этой модели (Appleton et al., 2007a).
OPEN QUESTIONS
Быстрые успехи человеческой и мышиной генетики в последние годы привели к идентификации нескольких новых транскрипционных регуляторов гипертрофии хондроцитов. это указывает на то, что гипертрофическая дифференцировка более сложная, чем предполагалось м нуждается во взаимодействии многих белков, потенциально способом. который зависит от анатомического места и/или специфической стадии развития. Одной из величайших задач будущего является расшифровка транскрипционных сетей, соединяющих описанные транскрипционные факторы (и несомненно др., которые будут идентифицированы) др. с др. и с в конечном итоге с генами эффекторами, такими как гены клеточного цикла и ECM. Большинство, но не все идентифицированные факторы, по-видимому, контролируют экспрессию Runx2 (Fig. 3), поэтому важно решить, какие из этих эффектов являются прямыми и какие непрямыми. Сходным образом интересно, как эти факторы взаимодействуют др. с др. - или путем перекрестной регуляции их экспрессии или благодаря прямым межбелковым взаимодействиям-всё это д. пролить свет на их роли в дифференцировке хряща. Анализ всего генома в отношении генов мишеней для каждого транскрипционного фактора д. быть осуществлен, используя как базирующиеся на РНК микромассивы в ходнроцитах с повышенной или пониженной функцией соотв. фактора (это не сможет различить между прямыми и непрямыми мишенями, но предоставит количественные данные по изменениям генной экспрессии) так и иммунопреципитацию хроматина в купе с геномными массивами ("ChlP-on-Chip," это сделает возможной идентификацию непосредственных белок-ДНК взаимодействий, но не даст информации, как ген мишень регулируется с помощью фактора связывания) Наконец, важно охарактеризовать пути, которые соединяют экспрессию и активность этих транскрипционных факторов со многими внеклеточными сигналами, контролирующими физиологию хондроцитов. Во всех этих экспериментах внимание д. быть уделено стадио- и location-специфическим эффектам как напр.. в случает дефектов, возникающих при мутациях SHOX у человека и Shox2 у мыши.
Др. не охарактеризованная область - это эпигенетическая регуляция дифференцировки хондроцитов. Упомянутый выше тяжелый скелетный фенотип Hdac4 нокаутных мышей предоставляет один из проблесков на роль таких эпигенетических механизмов, которые скорее всего играют роль в развитии хряща (Hug, 2004; Vega et al., 2004). Др. гистоновая деацетилаза, HDAC1, как было показано, взаимодействует с Nkx3.2 и обеспечивает транскрипционную репрессию с помощью этого белка (Kim and Lassar, 2003). Сходным образом инактивация хроматиновых белков HMGIM1 или HMGB1 влияет на дифференцировку хондроцитов (Furusawa et al., 2006; Taniguchi et al., 2007), a Runx2 и Sox9, как было установлено, взаимодействует с энзимами, участвующими в ремоделировании хроматина, такими как HDACs и p300 (Sierra et al., 2003; Schroederetal., 2004; Fur-umatsu et al., 2005; Westendorf, 2006; Jensen et al., 2008). Более того, мутации потери функции гена, кодирующего родственного p300 белок Creb-binding protein (CBP) у мышей (Tanaka et al., 1997; Oike et al., 1999) или при синдроме Rubinstein-Taybi у людей (Petrij et al., 1995) вызывают скелетные дефекты. Все вместе эти находки подчеркивают важную роль эпигенетических механизмов и процессов ремоделирования хроматина в развитии хряща в целом и особенно в функции обсуждаемых транскрипционных факторов.
Сайт создан в системе
uCoz
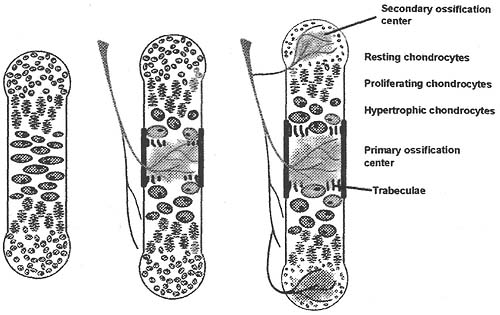 Figure 1. Endochondral ossification. During endochondral ossification, chondrocytes form a model of the later bones and form zones of resting, proliferating, and hypertrophic chondrocytes. Hypertrophic cartilage is invaded by blood vessels and ultimately replaced by bone tissue and bone marrow in the primary ossification center. Postnatally, a similar process results in the formation of the secondary ossification center, and the growth plate forms between primary and secondary ossification center. In humans, the growth plates close and the primary and secondary ossification centers fuse at the end of puberty.
Figure 1. Endochondral ossification. During endochondral ossification, chondrocytes form a model of the later bones and form zones of resting, proliferating, and hypertrophic chondrocytes. Hypertrophic cartilage is invaded by blood vessels and ultimately replaced by bone tissue and bone marrow in the primary ossification center. Postnatally, a similar process results in the formation of the secondary ossification center, and the growth plate forms between primary and secondary ossification center. In humans, the growth plates close and the primary and secondary ossification centers fuse at the end of puberty.