Мутантные мышиные модели, которые имеют HHL, обусловленные дефектами внутреннего уха, могут помочь идентификации генов, которые играют роль в развитии или функции внутреннего уха. Когда ген подозревается как ответственны за HHL у людей, то сходные мутации могут быть сконструированы у мышей, чтобы проверить гипотезу. Целенаправленный генный мутагенез или 'knockout' мыши также могут быть использованы для выяснения роли генов по сравнению с мышами дикого типа. Нокаутные мыши были получены для генов, подозреваемых как важные для слуха, благодаря известным взаимодействиям их продуктов с белками, кодируемыми др. известными генами, связанными с глухотой. или благодаря экспрессии их продуктов во внутреннем ухе. Кроме того, мышиные модели используются для идентификации новых генов, которые играют роль в развитии внутреннего уха и в нормальном слухе. Многие линии мышей с нарушениями слуха
возникли спонтанно в течение последнего столетия. Более того, соотв. мутагенез мышиных хромосом с помощью химических соединений (в основном с помощью ENU, N-ethyl-N-nitrosourea), рентгеновского облучения или соотв. инсерции чужеродных последовательностей ("gene trap") использовали для создания новых линий мышей с нарушениями слуха. Идентификация соотв. генов в таких линиях намного легче, чем анализ генетического сцепления у людей. Фактически, большинство генов, связанных с глухотой, было идентифицировано у людей только после их первоначальной идентификации у мышей с нарушениями слуха (Supplementary Table S1).
Мутации более чем 172 различных генов были описаны, как ответственные за нарушения или дисфункцию внутреннего уха у мышей (большинство из них приведено в Jackson Laboratory's Hereditary Hearing Impairment in Mice базе данных: http://www.jax.org/hmr/master_table.html). Лишь 44 из них уже были сцеплены с HHL у людей (эти гены представлены в Supplementary Table S1).
Кроме того, два гена, которые были сцеплены с HHL человека, оказались несущественными для развития и функции внутреннего уха у нокаутных мышей (Table 1). Figure 1 иллюстрирует пространственную экспрессию некоторых из белков, кодируемых генами, сцепленными с HHL.
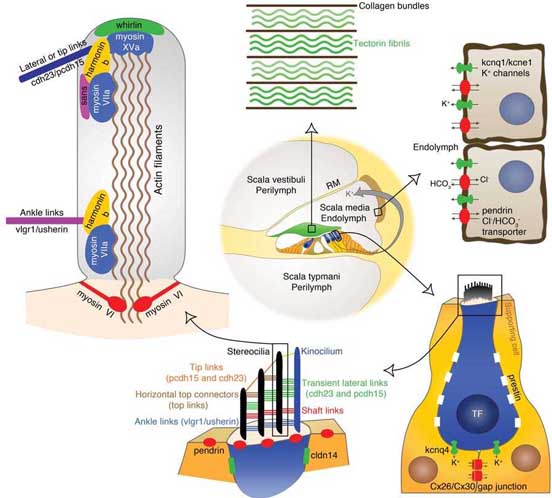
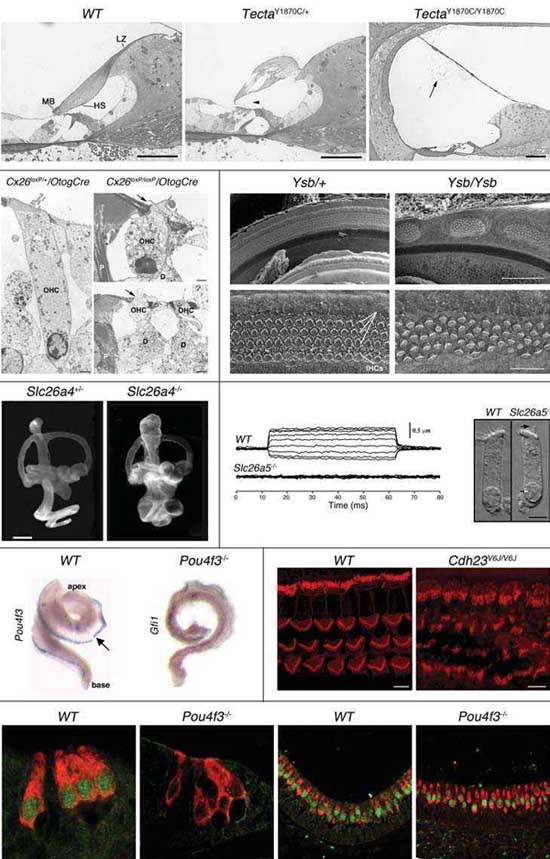
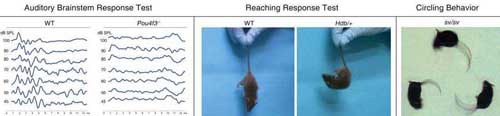
Вследствие идентификации мутантных генов, мутантные мыши могут быть использованы для отслеживания нарушений развития внутреннего уха и идентификации специфических ролей генных продуктов. Примеры методов, которые были использованы для оценки результатов мутаций, сцепленных с глухотой, показаны на Figures 2 и 3. Развитие и дефекты внутреннего уха можно отслеживать, используя световую микроскопию (Figure 2, A-C), трансмиссионную (TEM; Figure 2, D-F) или сканирующую электронную микроскопию (SEM; Figure 2, G-J), а также с помощью paintfill анализа (Figure 2, K-L). Физиологические подходы могут быть использованы для измерения токов ионов и потенциалов напряжения. Подход patch clamp может использоваться для измерения токов или мембранных потенциалов в одиночной клетке. Продолжительность изменения в индивидуальных клетках может быть использована для измерения electromotility наружных волосковых клеток (Figure 2, M-N). Временные и пространственные паттерны экспрессии специфических мРНК или белков в клетках могут наблюдаться с помощью in situ hybridization (ISH; Figure 2, O-P) и immunofluorescence (Figure 2, Q-V), соотв. Измерения auditory brainstem response (ABR) на звуковые сигналы с помощью электродов в скальпе широко используемый подход для оценки слуха у мышей (Figure 3, A-B). Вестибулярные дефекты могут быть оценены с помощью плаванья или др. поведенческих тестов и могут вызывать характерное поведение кружения (Figure 3, C-E).
репрезентативных генов, которые являются критическими для нормального развития и функции внутреннего уха млекопитающих. Мы сконцентрируемся в основном на на генах для внутриклеточных и интегральных белков внутреннего уха, которые были обнаружены, как участвующие в HHL людей и развитии внутреннего уха мышей. Тем не менее некоторые примеры генов, кодирующих внутриклеточные белки, также будут упомянуты.
Extracellular matrix components: cartilage and tectorial membrane defects (collagen genes and Tecta)
Синдром Stickler (Stickler et al., 1965) включает помимо прогрессивной сенсоронейральной HHL, преждевременные дегенеративные изменения в различных суставах с аномальным развитием эпифизов, аномалиями позвонков, остеоартритами и иногда также с необычным лицом и расщеплением нёба. Имеется 3 типа синдрома Stickler: тип 1 включает также прогрессивную миопатию и слепоту, обусловленную vitreoretinal дегенерацией и отслойкой сетчатки, тогда как тип 2 обладает др. дефектами стекловидного тела без отслойки сетчатки [rev. (Snead and Yates, 1999)]. Тип 3 является более легким, без миопатии и аномалий глаз (Vikkula et al., 1995). Сцепленные с синдромом Stickler коллагены Col2a1, Col11a1 и Col11a2 вляются важными компонентами не только TM улитки, но и хрящей (Col2a1 экспрессируется также в стекловидном теле глаза). Т.к. внутреннее ухо имеет хрящевое покрытие, которое играет важную роль в его эмбриогенеза, то мутантные коллагены типа II и XI влияют на размеры, структуру и развитие внутреннего уха.
Col9a1 является примером гена, который был связан с HHL у мышей ещё до его картирования как локуса, сцепленного с глухотой у людей. Col9a1-нокаутные мыши были получены в 1994, но внутреннее ухо у них не было изучено, а наблюдаемый фенотип характеризовался в основном не воспалительными заболеваниями суставов, напоминающими остеоартрит у людей (Fassler
et al., 1994). Лишь спустя 11 лет после установления, что Col9a1 экспрессируется на высоком уровне во внутреннем ухе людей (Abe et al., 2003) [коллагены IX были установлены как основной компонент TM
значительно раньше (Richardson et al., 1987)], стали исследовать внутреннее ухо нокаутных Col9a1 мышей (Asamura et al., 2005). В самом деле, эти мыши обнаруживали прогрессивную потерю слуха в основном из-за нарушений организации коллагеновых фибрилл в TM, что приводило к аномальной форме этой мембраны. TM у Col9a1 нокаутных мышей не содержит ни коллагенов IX, ни коллагенов II. Было предположено, что коллагены IX и II могут взаимодействовать в TM, чтобы предопределить её трехмерную структуру (Asamura et al., 2005; Suzuki et al., 2005). Годом позже мутация в COL9A1 была сцеплена с аутосомно рецессивным синдромом Stickler у людей (Van Camp
et al., 2006).
Cho мыши возникли спонтанно в 1971 (Seegmiller et al., 1971). Гомозиготы имеют расщепление нёба и погибают вскоре после рождения из-за летальной хондродисплазии. Мутация cho является делецией 1-nt в гене Col11a1, она вызывает сдвиг рамки считывания и образование кодона преждевременной терминации, что приводит к возникновению укороченного продукта, которые не может давать ансамбли с др. молекулами коллагенов. Т.о., cho по существу является функциональным нулевым аллелем Col11a1 (Li et al., 1995). Гомозиготы имели тяжелые нарушения слуха из-за недоразвития кортиева органа в нижнем витке улитки, с отсутствием волосковых и поддерживающих клеток, а также нервных окончаний и pillar клеток (Cho et al., 1991). Т.к. гетерозиготные cho мыши, которые экспрессировали и дикого типа и cho аллель Col11a1, страдали от зависимого от возраста остеоартрита, то было предположено, что аллель cho
может оказывать деструктивный эффект на соединительную ткань. Однако, гетерозиготные мыши слышали хорошо во время первых двух мес., но позднее у них развивалась умеренная и прогрессивная потеря слуха (зависимая от возраста), что недостоверно отличалось от мышей дикого типа (Szymko-Bennett et al., 2003). В противовес находкам у мышей, COL11A1-сцепленная SHL у людей проявлялась также у гетерозигот: точковая мутация в COL11A1 (G97V) была сцеплена с аутосомно доминантным синдромом Stickler (Richards et al., 1996) , а splice-donor-site мутация этого гена была сцеплена со сходным аутосомно-доминантным синдромом Marshall (Griffith et al., 1998).
Две мышиные модели с целенаправленными мутациями в Tecta (alpha tectorin) были получены той же самой группой. Обе мутации, индуцируют дефектные TM и HI. Первая мутация вызвана делецией в Tecta (наз. TectaΔENT). Единственным дефектом у гомозиготных мышей, которые не экспрессировали alpha tectorin (нулевая мутация), была TM, которая была лишена всего не-коллагенового матрикса и была полностью обособлена от кортиева органа и спиральной каймы (spiral limbus). Внутреннее ухо у них было менее чувствительным к стимуляции звуком, это подтверждает гипотезу, что TM умножает реакции волсковых клеток на сигналы низкого уровня (Legan
et al., 2000; Lukashkin et al., 2004). Проверка гомозиготных Tecta ΔENT/ΔENT мышей вместе с исследованиями движений TM и BM кортиева органа [e.g. (Hemmert et al., 2000)], помогло выявить роль этих мембран [см. (Legan et al. , 2005)]. Вторая мышиная модель, несущая missense мутации в Tecta (Legan et al., 2005), идентична мутации Y1870C, которая была обнаружена при нарушении слуха у людей (Verhoeven et al., 1998). Гомозиготные TectaY1870C/Y1870C имеют обособленную TM без tectorins, сходную с таковой мышей Tecta ΔENT/ΔENT. Гетерозиготные TectaY1870C/+ обладают разрушенной и частично истонченной TM которая экспрессируется tectorins и всё ещё частично соединена с кортиевым органом (Figure 2, A-C). Хотя взаимодействия между гетерозиготными TM и OHC выглядят нормальными, с почти нормальным транспортом обратной связи от OHC к BM, чувствительность к слуховым сигналам снижена из-за повышения порога нейральной активации. Пространство между TM и IHC увеличено у гетерозигот и движения IHC и reticular lamina специфически редуцированы в отношении частотных характеристик. Т.о., гетерозиготные TectaY1870C/+
помогли подтвердить вторую роль TM: хотя пучки волосков IHC не внедряются непосредственно в TM, TM всё ещё выполняет роль по трансмиссии вибраций BM к IHC с характерными частотами. Др. словами, TM приводит в порядок вибрации BM, чтобы оптимально стимулировать IHC при их наилучших частотах (Legan et al., 2005). Эта гипотеза недавно подтверждена с помощью физиологического исследования, подтвердившего, что пучки волосков IHC движутся в ответ перемещения жидкости в узком пространстве между IHC и TM. Эти движения жидкости результат вибрации TM и движений пучков волосков OHC (Nowotny and Gummer, 2006).
Если мутации в TECTA уже были сцеплены с NSHL у людей (Hughes et al., 1998; Verhoeven et al., 1998; Mustapha et al., 1999), то мутации TECTB topt не найдены у лиц с нарушениями слуха. Однако, нокаутные по beta-tectorin мыши недавно были получены. Хотя TM матрикс у гомозиготных мышей нарушен, их внутреннее ухо менее чувствительно только к тонам низки х частот, тогда как при высоких частотах разрешающая способность по частоте была обострена с незначительными или отсутствием нарушений чувствительности (sharpness cochlear tuning). Эти результаты указывают на третью роль TM: влиять на частотное разрешение улитки (Russell et al., 2007).
Intra-hair bundle link proteins: Cdh23, Pcdh15, Vlgr1
and Ush2a
Синдром Usher является наиболее распространенной этиологией комбинированной наследственной глухоты и слепоты. Это заболевание комбинирует врожденную сенсоронейральную потерю слуха и прогрессирующую потерю поля зрения из-за retinitis pigmentosa (RP), который ведет к прогрессирующей ретинальной дегенерации . Описаны три клинических субтипа синдрома Usher. Они отличаются началом и характером потери слуха, временем начала RP и вовлечением вестибулярной дисфункции [rev. (Nikolopoulos et al., 2006)]. Мутации 9 генов сцеплены с синдромом Usher у людей. 5 из этих генов сцеплены также с NSHL у людей: MYOVIIA, USH1C/Harmonin, CDH23, PCDH15 и VLGR1/MASS1 (Van Camp and Smith, 2006). Мутантные мыши сегодня получены по 8 генам, сцепленным с синдромом Usher. Белки, кодируемые генами Usher принадлежат разным классам и выполняют разные функции. Однако, все эти белки играют роль в молекулярной функции, развитии и/или поддержании пучков волосков волосковых клеток. Установлено, что все сцепленные с Usher белки связаны (непосредственно или косвенно) др. с др. посредством harmonin's PDZ сайтов и формируют мультибелковую единицу, которая может сновать (посредством моторных миозинов myosin VIIa и/или myosin XVa) вдоль актиновых филамент волосковых клеток к месту её действия в стереоцилиях (Figure 1F) [rev. (Kremer et al., 2006; Reiners et al., 2006)]. 4 Usher-сцепленных генов кодируют адгезивные белки (cadherin 23, CDH23; protocadherin 15, PCDH15; Very Large G-protein coupled Receptor-1, VLGR1/MASS1; и usherin, USH2A). Др. сцепленные с Usher- гены кодируют внутриклеточные белки волосковых клеток (Supplementary Table S1). Роль Usher-сцепленных белков недавно была рассмотрена в обзоре (Reiners et al., 2006).
Стереоцилии высоко специализированные микроворсинки с актиновым стержнем, которые проецируются из волосковой клетки в эндолимфатическое пространство. В пучках волосков стереоцилии ранжированы в ряды в виде специфического лестничного паттерна ( Figure 1, D-F). У мышей развитие пучков волосков происходит начиная с эмбриогенеза и заканчивая двумя первыми неделями после рождения. Usher-сцепленные адгезивные белки участвуют в связывании стереоцилий, что существенно для процесса механотрансдукции, с помощью которой волосковые клетки преддверия и улитки транслируют механические движения пучков своих волосков в электрохимические сигналы. Во внутреннем ухе млекопитающих развивающиеся и зрелые пучки волосков существенно отличаются (Figure 1E ).
Cadherin 23 (известный также как otocadherin, Cdh23) и protocadherin 15 (Pcdh15) являются трансмембранными белками с коротким внутриклеточным доменом и длинным внеулеточным доменом, которые являются атипическими членами сверхсемейства cadherin. Все cadherinмолекулы содержат cadherin домены ('EC' домены) вдоль своей внеклеточной части, которые обеспечивают Ca2+-зависимую димеризацию молекул cadherin. Димеризация кадхериновых белков от двух соседних клеток связывает эти клетки [rev. (Reiners et al., 2006)]. В своем цитоплазматическом конце, cadherin 23 и protocadherin 15 содержат class I-PDZ binding (PBM) мотивы, которые могут связывать PDZ-содержащие белки. Следовательно, они могут связывать harmonin. Посредством harmonin, сцепленные с Usher cadherins соединяются с цитоскелетными актиновыми филаментами и являются частью Usher-сцепленной мультибелковой единицы (Siemens et al., 2002; Adato et al., 2005b)].
Во внутреннем ухе мышей дикого типа, cadherin 23 локализуется на стереоцилиях волосковых клеток и Reissner's мембране (Wilson et al., 2001; Boeda et al., 2002; Lagziel et al., 2005). В пучках волосков мышей, cadherin 23 локализуется вдоль длины растущих стереоцилий и на кончиках зрелых стереоцилий. Точнее cadherin 23 располагается на связках между стереоцилиями пучков волосков (Boeda et al., 2002; Siemens et al., 2002; Siemens et al., 2004; Lagziel et al., 2005; Michel et al., 2005; Rzadzinska et al., 2005). Два сплайс-варианта Cdh23 найдены во внутреннем ухе мышей. Оба имеют PDZ-связывающие мотивы, которые связывают harmonin. Описан укороченный cadherin 23, который лишен внеклеточного домена [rev. (Reiners et al., 2006)]. Protocadherin 15 широко экспрессируется во многих тканях мышей (Alagramam et al., 2001a; Murcia and Woychik, 2001) и людей (Alagramam
et al. , 2001b), включая головной мозг, улитку и преддверие от раннего развития до взрослого периода. В развивающейся улитке protocadherin 15 располагается на апикальной поверхности волосковых и подерживающих клеток, клеток наружной борозды и клеток спирального ганглия, тогда как в зрелой улитке protocadherin 15 экспрессируется только в стереоцилиях волосковых клеток (Alagramam et al., 2001b).
Доступны многие мутантные по Cdh23 мыши. 4 разные мутации Cdh23 возникли спонтанно у мышей: waltzer (Deol, 1956; Di Palma et al., 2001a; Wilson et al., 2001; Lagziel et al., 2005), waltzer niigata (Wada et al., 2001), modifier of deafwaddler - mdfw (Bryda et al., 2001) и age-related hearing loss - Ahl (Noben-Trauth et al., 2003). С помощью chlorambucil индуцированы делеционные мутации [ Albany-waltzer (Bryda et al., 1997)] и с помощью ENU, вызывающего точковые мутации (получены три типа waltzer-Jackson аллелей; описаны в Mouse Genome Database: http://www.informatics.jax.org) как характерны для Cdh23 мутантных мышей. 7 из этих Cdh23-мутантных линий мышей (за исключением Ahl) обладают сходным фенотипом: NSHL с поведением вращения, качанием головы и беспорядочными движениями, которые появляются у гомозигот с рождения. Гетерозиготы выглядят нормальными при рождении, но обнаруживают тенденцию к прогрессивной потере слуха в более старом возрасте и имеют более высокую чувствительность к шумами вызываемой потере слуха (Holme and Steel, 2004). Аллель Ahl естественно возникший диморфизм Cdh23G753A, который возникает во многих широко распространенных лабораторных линиях мышей. Замещение guanosine 753 на adenosine вызывает in-frame пропуск экзона 7, приводящий к тенденции возникновения прогрессивной потери слуха во время старения и к высокой чувствительности индуцируемой шумами потери слуха (Davis et al. , 2001; Noben-Trauth et al., 2003).
Waltzer мутанты мышей обнаруживают прогрессивную дезорганизацию пучков волосков, которая впервые обнаруживается в начале формирования пучков на 18.5 день эмбриогенеза (E18.5) и становится наиболее выраженной в зрелых волосковых клетках (Figure 2, Q-R). Кроме того, kinocilium оказывается расположенным неправильно. В старшем возрасте стереоцилии кажутся толще и слиты, что ведет к дегенерации волосковых клеток (Di Palma et al., 2001a; Wada et al., 2001; Holme and Steel, 2002). C57BL/6J мыши, которые гомозиготны по аллелю Ahl обнаруживают дегенерацию волосковых клеток в старости, более выраженную в апикальной части улитки. OHC повреждаются больше, чем IHC. Наблюдается также дегенерация эфферентных нервных волокон (Mizuta
et al., 1993). В развивающихся волосковых клетках внутреннего уха мышей cadherin 23 располагается как на kinocilial, так и на временных боковых связках (Boeda et al., 2002; Lagziel et al., 2005; Michel et al., 2005), but waltzer мутантный cadherin 23 отсутствует только в латеральных связках. Cadherin 23 обнаруживается также вдоль kinocilia зрелых вестибулярных волосковых клеток (Lagziel et al., 2005). Волосковые клетки Cdh23-дефицитных мутантов рыбок данио лишены кончиковых связок и эти рыбки имеют дефекты баланса и слуха (Sollner et al., 2004). Две группы описали, что cadherin 23 у мышей также является компонентом кончиковых связок между стереоцилиями пучков в улитке и преддверии. Более того, cadherin 23 обладает биохимическими свойствами, сходными с теми, что характерны для кончиковых связок. Следовательно, было предположено, что cadherin 23 составляет кончиковые связки, которые регулируют механически закрываемые-открываемые ионные каналы в стереоцилиях волосковых клеток (Goodyear and Richardson, 2003; Siemens et al., 2004).
Первая мышиная модель мутантного Pcdh15 была Ames-waltzer (av). Первоначально мыши Ames-waltzer были описаны в 1956 как несущие спонтанную рецессивную мутацию, вызывающую глухоту, поведение кружения, трясения головой и гиперактивности, сходными с waltzer ( v) фенотипом (Schaible, 1956). В последующие годы несколько мутаций того же самого локуса возникли независимо, давая сходные фенотипы. Мутантный ген был обнаружен как Pcdh15 аллель Ames-waltzer, который возникает у трансгенных мышей вследствие инсерционного мутагенеза (Alagramam et al., 1999). Поведение кружения и пониженное потребление краски AM1-43, которое, как было установлено, коррелирует с нормальной функцией трансдукции в волосковых клетках предшествовало структурным дефектам в преддверии, что можно было наблюдать в световом и электронном микроскопе. Функциональный дефект ведет к дезорганизации стереоцилий улитки и saccule, что приводит к дисфункции волосковых клеток и прогрессивной дегенерации. Если во внутреннем ухе на ст. P10 гомозиготы обнаруживают только аномальные стереоцилии в улитке, то saccular стереоцилия начинают дезорганизовываться только на P30 , а во внутреннем ухе взрослых гомозиготных мышей (P50 или старше) обнаруживается практически полная дегенерация кортиевого органа улитки и saccular macula преддверия (отсутствуют и поддерживающие и волосковые клетки). В улитке наблюдается вторичная дегенерация нейронов спирального ганглия. Нейроэпителий гребней utricle и полукружных каналов выглядит нормально. но utricular отолиты крупные и уродливые (Alagramam et al., 1999; Alagramam et al., 2001a; Alagramam et al., 2005). У др. спонтанного Pcdh15 мутанта, возникшего в результате инсерции остатка cytosine, что привело к сдвигу рамки считывания и преждевременному стоп-кодону, фенотипу, сходному, хотя мыши не были полностью глухи. Дезорганизация стереоцилий улитки наблюдается у новорожденных (P0) (Hampton et al. , 2003). ENU-индуцированные Pcdh15 мутантные мыши дают сходный фенотип с дезорганизацией стереоцилий улитки, но не раньше P2. В улитке IHC были менее повреждены, чем OHC (Washington et al., 2005). Три модели, описанные выше являются гомозиготами по функциональным нулевым аллелям. Более умеренные фенотипы описаны у мышей, гомозиготных по менее тяжелым мутациям Pcdh15 (Pawlowski et al., 2006).
В волосковых клетках внутреннего уха мышей несколько изоформ protocadherin 15 экспрессируются и две из них, как полагают, являются частью комплексов кончиковых и kinocilial связок в пучках волосков. Др. изоформа может быть ассоциирована с временными боковыми связками между развивающимися стереоцилиями и с kinocilial связками, т.к. паттерн экспрессии этой изоформы сходен с таковым для cadherin 23 (Ahmed et al., 2006). Ген Vlgr1/Mass1 у мышей транскрибируется в несколько сплайс-вариантов, которые кодируют интегральные и секретируемые белки. Самая длинная изоформа Vlgr1b, которая приблизительно в 19 kb, транслируется в самый большой из известных белков клеточной поверхности (~ 6300 аминокислот), содержащий крупный внеклеточный домен. Его внутриклеточный домен содержит PBM мотив, который может взаимодействовать с harmonin's PDZ доменом. Хотя белок Vlgr1b обладает типичной структурой G-protein coupled рецептора с 7 трансмембранными доменами, его функция неизвестна (McMillan
et al., 2002; Yagi et al., 2005). Mass1 является самым маленьким (~ 9400 оснований ) сплайс-вариантом Vlgr1. Vlgr1 рецепторы экспрессируются преимущественно в нейроэпителии развивающегося головного мозга мышей (Yagi et al., 2005) Ю а мутации Vlgr1, в особенности Vlgr1b и
Mass1 транскрипты, ассоциируют с аудиогенными судорогами у мышей (Skradski et al., 2001; McMillan and White, 2004; Yagi et al., 2005) и судорогами у людей (Nakayama et al., 2002). Внеклеточные домены Vlgr1 рецепторов содержат множественные повторяющиеся единицы CalX-модулей, которые соединяются с катионами Ca2+ и могут играть роль в Ca2+-зависимой межклеточной адгезией. Было предположено, что эти модули могут отслеживать уровни внеклеточного Ca2+ и участвовать во внутри- и внеклеточном переносе Ca2+ (Nikkila et al., 2000; Weston et al., 2004). Дополнительные мотивы во внеклеточных доменах Vlgr1 белков, как полагают, взаимодействуют с др. Usher-сцепленными белками [rev. (Reiners et al., 2006)].
Первой мышиной моделью мутантов Vlgr1 была Mass1Frings , которая возникла случайно в 1951 (Frings et al., 1951), она служила мышиной моделью эпилепсии, обусловленной чувствительностью к громкими шумами вызываемым судорогам. BUB/BnJ инбредная линия мышей является гомозиготной по Mass1Frings мутации и обнаруживает как аудиогенные судороги, так и прогрессивную потерю слуха, которая начинается постнатально и прогрессирует до полной глухоты (Zheng et al., 1999; Skradski et al. , 2001). BUB/BnJ мыши были гомозиготными по Ahl аллелю Cdh23, но этот факт не объясняет глухоты всех этих мышей, т.к. др. линии гомозигот по Ahl по вероятности и тяжести потери слуха намного ниже. Ассоциация мутаций VLGR1 с HHL, индуцируемой при Usher type II синдроме у людей (Weston et al., 2004) открывает возможность, что мутация Mass1Frings лежит в основе потери слуха у BUB/BnJ мышей. В самом деле, было показано, что ко-мутации Cdh23 и Vlgr1 is ответственны за наиболее тяжелую потерю слуха у BUB/BnJ мышей. У молодых BUB/BnJ мышей стереоцилии улитки развиваются аномально и остаются незрелыми. Стереоцилии рассоединены и обособлены, иногда обнаруживаются вне своих единиц, наиболее тяжело поврежденныее пучки теряют свою полярность и градированную высоту. У старых волосковые клетки и клетки спирального ганглия дегенерируют (Johnson et al., 2005).
Экспрессия дикого типа Vlgr1 рецепторов во внутреннем ухе ограничена регионом синапсов и стереоцилиями волосковых клеток в преддверии и улитке. В волосковых клетках Vlgr1 рецепторы экспрессируются только в основании развивающихся стереоцилий в той же самой локализации и в то же время, что и ankle связки: их экспрессия максимальна в перинатальный период и уменьшается во время развития волосковых клеток. Моноклональные антитела, которые используются для идентификации ankle связок у кур, как было установлено, соединяются с птичьим ортологом Vlgr1b. Получены две мышиные модели и мутантным Vlgr1: (a) нокаутные мыши, которые не экспрессируют Vlgr1 белков (Yagi et al., 2005) и (b) Vlgr1/del7TM мыши, у которых были использована адресная делеция, чтобы удалить трансмембранный домен Vlgr1 (McGee et al., 2006). В обеих моделях, дефицит рецепторов Vlgr1 receptors приводил к сходным аномалиям улитки. Гомозиготные мыши не имели ankle связок между стереоцилиями волосковых клеток. Хотя пучки волосков выглядели нормально при рождении, затем они становились дезорганизованными. Мыши, гомозиготные по мутантному Vlgr1 обнаруживали выраженную глухоту на 3-ей неделе жизни и с этого возраста обнаруживали дезорганизованные пучки волосков, включая смещение киноцилия, что приводило к развитию искривленных стереоцилий. Т.о., Vlgr1 рецептор, как полагают. является критическим для комплекса ankle связок. Неожиданно, хотя обычно развивающиеся вестибулярные пучки волосков имели ankle связки и экспрессировали также Vlgr1 , только волосковые клетки улитки были повреждены у гомозиготных мышей. Вестибулярные клетки не дегенерировали и не наблюдалось фенотипических отклонений в преддверии, что согласуется с отсутствием вестибулярных симптомов у пациентов с Usher II (McGee et al., 2006; Yagi et al., 2007).
Др. интегральныйl Usher-сцепленный бело, usherin (кодируемый длинным транскриптом Ush2a), как полагают, также является компонентом ankle связок в развивающихся стереоцилиях (Adato et al.,2005a). У людей мутации USH2A ответственны за наиболее распространенную генетическую форму синдрома Usher (Eudy et al., 1998). Сходным образом, в то время как нокаутные Ush2a-/- мыши обнаруживают прогрессивную дегенерацию фоторецепторных клеток, их слух повреждается умеренно, характеризуя легкую не прогрессирующую HI на высоких частотах. Хотя usherin, как полагали, является частью ankle белка и обнаруживается в основном в основании развивающихся стереоцилий как внутренних, так и наружных волосковых клеток (со ст. E20) вдоль всей улитки, Ush2a-/- мыши имеют нормальные пучки волосков и теряют только OHC в базальном витке улитки (Adato et al., 2005a; Liu et al., 2007).
Исследование, рассмотренные выше подтверждают, что экспрессия молекул связок в раннем развитии пучков волосков является существенной для правильного их образования и созревания. Корректное созревание пучков волосков является критическим для жизнеспособности волосковых клеток.
Genes responsible for endolymph production
Улитка содержит два разделенные, заполненные жидкостью компартмента с разными концентрациями ионов (Figure 1A). Перилимфатическое пространство содержит перилимфу, раствор с высоким содержанием Na+ и низким K+ , сходный с др. внеклеточными жидкостями тела. Верхушки волосковых клеток обращены лицом в эндолимфу, которая имеет противоположный состав катионов, высокий K+ и низкий Na+, тогда как их базолатеральная поверхность омывается перилимфой. Циркуляция ионов калия в улитке из перилимы в эндолимфу посредством боковых стенок улитки и поддержание уникального состава ионов в эндолимфе являются существенными для функции слуха. Исследования мышных моделей, мутантных по белкам, участвующим в рециклинге K+ в улитке помогло установить механизм повторного использования. Вызываемые звуками потенциалы рецепторов, генерируются за счет притока ионов K из эндолимфы в волосковые клетки. Эти K+ ионы затем секретируются базолатерально во внеклеточное пространство кортиевого органа и подбираются поддерживающими клетками. Затем K+ ионы транспортируются латерально в направлении спиральной лигаменты посредством щелевых соединений между поддерживающими клетками и из поддерживающих клеток в корешковые (root) клетки, высвобождается во внеклеточное пространство спиральной лигаменты и затем с помощью вторичной сети щелевых соединений между фиброцитами соединительной ткани катионы переносятся в stria vascularis. K+ ионы проходят базальную мембрану между соединительной тканью и эпителиальными клетками stria vascularis посредством плотных соединений и высвобождаются из эпителиальных базальных клеток во внеклеточное пространство stria vascularis. Затем маргинальные клетки stria vascularis накапливают ионы K+ и высвобождают их обратно в эндолимфу. Маргинальные клетки stria vascularis и Deiters' клетки, как пример поддерживающих клеток, показаны на Figures 1C и 1D, соотв. Сходный путь рециклинга существует и в преддверии. Это описание несколько упрощено, т.к. происходит некоторое протекание K+ из эндолимфы через клетки наружной бороздки и Reissner's мембрану [rev. (Kikuchi et al., 2000; Wangemann, 2002)].
Несколько генов, которые объясняют HHL у людей, кодируют белки, которые участвуют в циркуляции K+ в улитке. Мутантные мышиные модели были разработаны для след. генов: (a) Gjb2/Cx26, Gjb6/Cx30 и Cldn14, которые кодируют межклеточные адгезивные белки: Gjb2 и Gjb6 гены кодируют белки щелевых соединений connexin 26 (Cx26) и connexin 30 (Cx30), тогда как Cldn14 кодирует белок плотных соединений; (b) Kcne1, Kcnq1 и Kcnq4, которые кодируют каналы калиевых ионов; и (c) Slc26a4 , который кодирует транспортер анионов. Щелевые соединения и каналы обеспечивают взаимосвязь двух клеток и делают возможным быстрый транспорт широкого разнообразия ионов и малых молекул (включая нуклеотиды, siRNAs и inositol phosphates) между соединенными клетками. Щелевые соединения состоят из тесно аггрегированных внутримембранных частиц каналов (connexons), которые в свою очередь являются гексамерными ансамблями конексиновых белков. Две самостоятельные сети щелевых соединений существуют в улитке: между клетками соединительной ткани и между не-сенсорными эпителиальными клетками. Cx26 и Cx30 являются частью обеих систем щелевых соединений и могут собираться совместно, чтобы сформировать гибридные (гетеромерные) щелевые соединения. Однако, превалирующая изоформа коннексинов, экспрессируемой в поддерживающих клетках улитки, это Cx26 (Ahmad et al., 2003; Forge et al., 2003; Buniello et al., 2004). В геноме человека гены GJB2 и GJB6 локализуются в том же самом хромосомном локусе ( DFNB1, 13q11-12). Мутации в этом локусе объясняют высокую пропорцию врожденной наследственной NSHL с вариабельностью, зависящей от популяции [~30-60%; (Zelante et al., 1997)]. GJB2 мутации являются наиболее превалирующим источником наследуемой глухоты у людей (30-50% из prelingual наследственной NSHL). В большинстве этих случаев ответственными мутациями являются небольшие делеции в гена GJB2 с аутосомно рецессивным типом наследования. Однако, немногие случаи доминантно наследуемых SHL, обуславливаемые мутациями GJB2, также описаны. Т.о., более сотни связанных с глухотой различных мутаций в GJB2 идентифицировано у людей. Крупные делеции в гене GJB6 также могут вызывать глухоту у гомозигот. Кроме того, комбинация крупной делеции в GJB6 и точковой мутации и GJB2 могут индуцировать NSHL у гетерозигот [Connexins and Deafness Homepage; http://davinci.crg.es/deafness/ (Ballana et al., 2007)].
Два разных подхода целенаправленного мутагенеза (Gabriel et al. , 1998) и ENU-индуцированный мутагенеза (Coghill et al., 2002), были использованы для нокаута Gjb2/Cx26 гена у мышей. Оба подхода привели к появлению хорошо слышащих гетерозигот, тогда как гомозиготные эмбрионы погибали in utero из-за дефектов плаценты. Были разработаны две дополнительные стратегии получения мутантных Gjb2 модельных мышей, которые были бы жизнеспособны и имели дефекты слуха. Gjb2 подвергнут специфическому нокауту в эпителиальной сети улитки (поддерживающих и фланкирующих эпителиальных клетка), используя условную систему cre-loxP, чтобы получить мышей, гомозиготных по Gjb2-loxP и несущих Cre после Otog промотора, который экспрессируется только в эпителиальных клетках улитки (Figure 2, D-F) (Cohen-Salmon et al., 2002). При втором подходе, целенаправленный точечный мутагенез был использован для репликации мутации Cx26 R75W (Kudo et al., 2003), которая ответственна за аутосомно доминантную SHL (HHL и болезнь кожи) у гетерозиготных пациентов (Richard et al. , 1998). Доминантное наследование объясняется способностью мутантов Cx26 ингибировать функцию щелевых соединений, которые собирают вместе дикого типа и мутантные Cx26 молекулы (Richard et al., 1998).
как Gjb2 нокаутные гомозиготы, так и Cx26 R75W гетерозиготы давали сходные HI у взрослых и гистологические фенотипы, хотя вторая модель давала более тяжелый фенотип. В обеих моделях развитие внутреннего уха было нормальным до постнатального дня 14 (P14). Лишь после начала восприятия звуков на P15-P16, эпителиальные клетки начинали погибать от апоптоза. Соседние с IHC поддерживающие клетки повреждались первыми. Затем OHC и их поддерживающие клетки начинали погибать. Кортиев канал спадался. Cx26 R75W гетерозиготы обнаруживали дегенерацию всего органа Корти, которая начиналась на ст. P14 и вела к полной дегенерации как волосковых, так и поддерживающих клеток в возрасте 7 недель. У Gjb2 нокаутных мышей, IHC погибали только у мышей с наиболее выраженной потерей слуха (но имели незрелые синапсы, даже если они выживали) и некоторые intradental клетки спирального limbus дегенерировали у старых мышей (P60). Ретикулярная lamina на апикальной поверхности сенсорного эпителия, которая представлена плотными соединениями между волосковыми клетками и их поддерживающими клетками, разрушена начиная с ранней стадии у Gjb2 нокаутных мышей (Figure 2, E-F). Следовательно, Cx26, по-видимому, важен для жизнеспособности и функционирования кортиева органа, но не нужен для его нормального развития. Различия между моделями были обнаружены в поддержании различий электрического потенциала между эндолимфатическим и перилимфатическим компартментами улитки, характеризуемого endocochlear potential (EP). У Gjb2 нокаутных мышей эндолимфатическая концентрация K+
и EP были более низкими у гомозиготных мышей, как и ожидалось, что подтверждает гипотезу, что базирующиеся на Cx26 щелевые соединения необходимы для рециклинга K+ в улитке. Неожиданно EPs Cx26 R75W были нормальными, указывая тем самым, что причиной апоптоза клеток в кортиевом органе в присутствии мутации Cx26 является нарушение транспорта K+ поддерживающими клетками скорее, чем нарушением гомеостаза эндолимфы, как первоначально предполагалось. Т.к. Cx26 не подвергся нокауту в преддверии в conditional модели и его экспрессия в преддверии была нормальной у гомозиготных мышей, то эти мыши не обнаруживали вестибулярных дефектов. Однако, ни вестибулярных и ли др. аномалий не было обнаружено во второй модели также. Кроме того, хотя доминантный мутант Cx26 R75W экспрессировался также в клеточной системе соединительной ткани улитки , не наблюдалось очевидных структурных изменений в stria vascularis или spiral ligament (Cohen-Salmon et al., 2002; Kudo et al., 2003).
Модельные Gjb6 нокаутные мыши были также получены с помощью инсерции миссенс мутаций. Гомозиготные мыши были жизнеспособны и плодовиты, но слух у них нарушен и отсутствовал EP. Дегенерация кортиева органа, обусловленная апоптозом, наблюдалась с возраста P18, одинаково с мутантными Gjb2 мышами (Teubner et al., 2003). Cx26 и Cx30 собираются вместе в одних и тех же щелевых соединениях (Ahmad et al., 2003; Forge et al., 2003). Хотя Cx30 не способен формировать гомомерные щелевые соединения в Cx26-дефицитных клетках, Cx30 не может компенсировать отсутсвие Cx26 в условной нокаутной модели (Cohen-Salmon et al., 2002).Разные коннексины отличаются поразмеру и ионной избирательности и имеют разную voltage-gating чувствительность. Как результат, connexons, собираемые из разных коннексинов, имеют разные фильтрующие и пропускные функции (Bruzzone and Cohen-Salmon, 2005; Zhao et al., 2006)]. Т.о., гомомерные коннексоны, собираемые тольк из Cx30 могут отличаться от подобных гетромерных коннексонов, собираемых из Cx26 и Cx30. Даже если проницаемость для малых ионов (подобных K+)
одинакова в разных типах коннексонов, доставка крупных молекул вторичных мессенджеров может быть отличной, затрагивающей косвенно приток K+. Недавно установлено, что Gjb2 мутации затрагивают проницаемость щелевых соединений для inositol triphosphate скорее. чем для K+ . Неспособность повторно использовать K+ из поддерживающих клеток обратно в эндолимфу является вторичной, как было установлено, по отношению к транспорту inositol triphosphate (Beltramello et al., 2005). Тем не менее, неспособность Cx30 компенсировать отсутствие Cx26 может быть результатом его низкой экспрессии. В противном случае избыточная экспрессия Cx26 у Gjb6 нокаутных мышей полностью восстанавливает слуховую чувствительность и предопределяет дегенерацию волосковых клеток. Т.о., по крайней мере, Cx26 может компенсировать отсутствие Cx30, подтверждая тем самым, что гетеромерные щелевые соединения, которые содержат Cx26 и Cx30 не существенны для нормального слуха и для жизнеспособности кортиевого органа у мышей. Интересно, что Gjb6 нокаутные мыши экспрессируют низкие уровни Cx26 белка в улитке, указывая тем самым на ускоренную деградацию гомомерных щелевых соединений. Gjb6 нокаутные мыше, которые также несут ген с избыточной экспрессией Cx26, избыточно экспрессриуют Cx26 в печени, но не в улитке, где уровни Cx26 нормальные, значит гомомерные Cx26 щелевые соединения менее стабильны, чем гетеромерные Cx26-Cx30 ансамбли, но имеют сходные функции (Ahmad et al., 2007).
Хотя connexin 29 (Cx29) не участвует в рециклинге K+ ионов в улитке, он заслуживает снимания, т.к. мутации в гене GJE1/Cx29 были обнаружены у пациентов с NSHL (Yang et al. , 2007). Распределение Cx29 в улитке очень отличается от такового для Cx26 и Cx30. В отличие от Cx26 и Cx30, которые в основном экспрессируются в поддерживающих клетках и фиброцитах улитки, Cx29эк в основном в Шванновских клетках спирального ганглия и в меньшей степени в stria vascularis (Eiberger
et al., 2006; Tang et al., 2006b). Экспрессия Cx29 в головном мозге и др. органах в основном касается миэлинизированных клеток. Две группы получили нокаутных мышей Gje1. Одна группа сообщила от отсутствии аномалий у Cx29-дефицитных C57BL/6 мышей, , включая нормальные миэлиновые слои (Eiberger et al., 2006), др. группа сообщила от потере слуха вследствие тяжелой димиэлинизации тел нейронов спирального ганглия (neuropathy), с пенетрантностью ~50% и не обнаружила повреждений в нейроэпителии внутреннего уха у мышей BALB/c (Tang et al., 2006b).
Плотные соединения большинства апикальных соединений в эпителиальных клетках служат в качестве главного ион-фильтрующего барьера против околоклеточного переноса жидкости. Кроме того, они вносят вклад в поддержание клеточной полярности путем образования внутримембранного барьера, который ограничивает латеральную диффузию компонентов апикальной и базалотеральной частей мембраны. Плотные соединения состоят из, по крайней мере, трех типов трансмембранных белков: occludin, claudins и членов семейства junction adhesion molecule (JAM). Известно более 20 claudins, каждый с характерной проницаемостью [rev.(Kondoh et al., 2006)]. В улитке важно отделение перилимфы от эндолимфы, которое обеспечивается с помощью плотных соединений, которые заполнят пространства между клетками, ограничивая компартменты жидкостей. После идентификации рецессивных мутаций CLDN14, как отвечающих за выраженную NSHL у людей (Wilcox et al., 2001), были получены Cldn14-нулевые мыши, для изучения роли claudin 14 во внутреннем ухе. Claudin 14 был обнаружен в плотных соединениях reticular lamina улитки (плотные соединения между волосковыми клетками и поддерживающими клдетками и между соседними поддерживающими клетками). Cldn14-нулевые мыши имеют нормальный EP, но глухи. Не обнаруживаются фенотипические отклонения в преддверии. Хотя плотные соединения reticular lamina выглядят микроскопически нормальными у Cldn14-нулевых мышей, стереоцилии волосковых клеток были потеряны или дезорганизованы во время первых трех недель жизни, что быстро сопровождалось дегенерацией волосковых клеток. OHC дегенерировали раньше IHC. Т.к. claudin 14 обладает более высокой проницаемостью для K+, чем для Na+ , он может быть необходим для поддержания собственно ионного состава перилимфатической жидкости, окружающей базолатеральную поверхность OHC. Правильный ионный состав этой жидкости может быть существенным для жизнеспособности OHC (Ben-Yosef et al., 2003).
Гены Kcne1, Kcnq1 и Kcnq4 кодируют субъединицы slow voltage активируемых калиевых каналов, которые являются основными детерминантами клеточной реполяризации в возбудимых клетках. Они открываются в ответ на деполяризацию и облегчают избирательный отток K+ через плазматическую мембрану. Каждый канал состоит из 4-х alpha нескольких beta субъединиц. Т.к. формирующие пору alpha субъединицы достаточны для образования функциональных каналов, то beta
субъединицы предопределяют уникальные свойства каналов, включая их одноканальную проводимость, общую активность канала, voltage зависимость, зависимость от времени активации, чувствительность к температуре и pH, а также чувствительность к лекарствам [rev. (Wangemann, 2002)].
Маргинальные клетки stria vascularis и вестибулярные темные клетки секретируют K+ в эндолимфу только с помощью K каналов, состоящих из Kcnq1 (alpha) и Kcne1 (beta) субъединиц. Следовательно, Kcnq1/Kcne1 каналы ответственны за формирование эндолимфы (Marcus et al., 1997; Neyroud et al., 1997; Marcus et al., 1998; Nicolas et al., 2001). В кардиальных миоцитах Kcnq1/Kcne1 K+ каналы несут медленно активирующийся rectifier тока K+, который играет основную роль в фазе реполяризации кардиального потенциала действия. Следовательно, мутации в
KCNE1 или KCNQ1 у людей индуцируют неразличимые SHL фенотипы (Jervell and Lange-Nielsen Syndrome) HHL и кардиальные симптомы, включая удлиненный QT интервал и аритмии, сопровождаемые обмороками или внезапной гибелью (Neyroud et al., 1997; Schulze-Bahr et al., 1997; Tyson et al., 1997).
Kcne1 (Vetter et al., 1996; Nicolas et al., 2001) или Kcnq1 (Lee et al., 2000; Casimiro et al., 2001; Rivas and Francis, 2005) нокаутные мыши обладали классическим waltzer-подобным фенотипом с тяжелой потере слуха и вестибулярными симптомами, вплоть до полной глухоты у взрослых мышей. Хотя гистология внутреннего уха была нормальной при рождении, изменения возникали позже. Strial маргинальные клетки и вестибулярные темные клетки были неспособны секретировать ионы K
+ , что приводило ко вторичной дегенерации нейроэпителия, включая волосковые клетки и к коллапсу эндолимфатического пространства. Сходным образом эндолимфатическое пространство спадалось у пациентов с синдромом Jervell и Lange-Nielsen (Friedmann et al., 1966). При рождении у мышей дикого типа type EP очень низок с высокими Na
+ и низкими K
+ концентрациями в эндолимфе. После рождения EP постепенно увеличивался (особенно со ст. P7), достигая взрослых значений к P14 (Yamasaki et al., 2000). Соотв., Kcne1 (Vetter et al., 1996) или Kcnq1 (Casimiro et al., 2001) нокаутные мыши обладали нормальным эндолимфатическим пространством при рождении. Только спустя 3 днф после рождения происходил коллапс Reissner's мембраны и уменьшение объема эндолимфатического пространства становилось видимым. Спонтанная точковая мутация в Kcne1 также возникала у мышей (
punk rocker мыши; Kcne1 pkr). Гомозиготные мыши, экспрессирующие сильно укороченный Kcne1 белок имели сходный фенотип с тем, что у Kcne1 нокаутных мышей (Letts et al., 2000). Kcnq1 нокаутные мыши также обнаруживали дефекты кардиальной реполяризации (Casimiro et al., 2001; Casimiro et al., 2004). В то время как Kcnq1 является сердцевиной канала, очевидно, что Kcne1 необходим для его доставки в плазматическую мембрану, т.к. вестибулярные темные клетки у Kcne1 нокаутных мышей экспрессировали Kcnq1 в своей цитоплазе скорее, чем в своих апикальных мембранах (Nicolas et al., 2001). Т.о., Kcne1 , по-видимому, является существенным для доставки Kcnq1 в мембрану и/или стабильности Kcnq1 в мембране. Kcnq4 является alpha субъединицей M-type K
+ канала. M-типа каналы являются очень медленными voltage-dependent K
+ каналами. В нейронах M-каналы могут противостоять неугасающей деполяризации мембраны и повторяющемуся возбуждению потенциала действия вследствие сильного возбуждающего сигнала, но они также могут временно повышать возбудимость нейронов вследствие воздействия на них модулирующих нейротрансмиттеров (Cooper and Jan, 2003). Соотв., Kcnq4 каналы были обнаружены в нейронах некоторых ядер центрального слухового пути. Однако, Kcnq4 обнаружен также в базолатеральных частях мембраны волосковых клеток улитки (Beisel et al., 2000) и преддверия (Rocha-Sanchez et al., 2007) мышей ( OHC и IHC). После начала восприятия слуха (P12-14),он локализуется исключительно на базальном полюсе. Поэтому было предположено, что Kcnq4 каналы ответственны за секрецию излишка ионов K
+ из волосковых клеток в перилимфу, окружающую их базолатеральную мембрану, и за установку потенциала покоящейся мембраны в волосковых клетках (Kharkovets et al., 2000; Boettger et al., 2002; Beisel et al., 2005; Rocha-Sanchez et al., 2007). У людей KCNQ4 мутации вызывают аутосомно доминантную NSHL, указывая тем самым, что мутантный ген оказывает доминантный, негативный эффект, если ко-экспрессируется с аллелем дикого типа (Kubisch et al. , 1999). Две мышиные модели с мутантными Kcnq4 были получены: гомозиготные нокаутные мыши и knock-in мыши с точковой мутацией, которая имитирует доминантно негативную мутацию у людей. Не выявлено вестибулярных симптомов в обеих мышиных моделях, хотя Kcnq4 строго экспрессируется в WT вестибулярных волосковых клетках. Мыши имеют нормальный слух на постнатальных стадиях, но обнаруживают прогрессирующую потерю слуха, которая сопровождается прогрессивной дегенерацией OHC. Прогрессирование как глухоты, так и потери OHC было более быстрым у гомозиготных нокаутов и knock-in мышей (недели) по сравнению с гетерозиготными knock-in мышами (месяцы). Использовав селективный ингибитор Kcnq каналов для выделения Kcnq-зависимых токов K
+ , не выявили Kcnq-зависимых K
+ токов в OHC от гомозиготных или доминантно негативных гетерозиготных мышей, это давало в результате потенциалы деполяризованной покоящейся мембраны OHC. IHC практически не затрагивались. Следовательно, было предположено, что мутации Kcnq4 индуцируют прогрессирующую HHL в результате хронической деполяризации OHC, ведущей к их дегенерации (Kharkovets et al., 2006). Недавно, экспрессия Kcnq4 в OHC , как было установлено, регулируется с помощью тироидных гормонов. Рецепторы тироидного гормона TR непосредственно затрагивают экспрессию Kcnq4 во время финальной дифференцировки OHC. У TR 1 нокаутных мышей, Kcnq4 экспрессируется, но аномально распределяется вдоль базальной и латеральной честей мембраны OHC (Winter et al., 2006).
Семейство SLC26 (solute carrier protein 26) обменников анионов включает интегральные белки с 10-12 трансмембранными доменами, которые могут транспортировать некоторые анионы, включая chloride, iodide, sulfate, nitrate, bicarbonate, hydroxyl, oxalate и formate. Каждый член этого семейства имеет отличное сродство и специфичность в отношении каждого аниона. Два члена SLC26 оказались сцеплены с HHL у людей: SLC26A4/pendrin и SLC26A5/prestin. Мутации SLC26A4 были ассоциированы как с SHL (Pendred syndrome) (Everett et al., 1997) , так и NSHL (Li et al., 1998; Usami et al., 1999), тогда как SLC26A5 оказался ассоциированным только с NSHL (Liu et al., 2003). Pendred Syndrome, впервые был описан в 1896 (Pendred, 1896), он характеризуется сенсоронейральной глухотой и увеличенным тироидным желваком с повышенным испусканием iodine после воздействия perchlorate. Большинство пациентов также обнаруживают радиологически обнаружимые структурные аномалии внутреннего уха, наиболее распространенным признаком корорых является увеличенный вестибулярный эндолимфатический канал [rev. (Glaser, 2003)]. Увеличенные эндолимфатические каналы наблюдаются также у некоторых пациентов с NSHL, обусловленными мутациями SLC26A4 (Li et al., 1998; Usami et al., 1999). В системах гетерологичной экспрессии pendrin , как было установлено, транспортирует iodide, chloride, formate и nitrate (Scott et al., 1999; Scott and Karniski, 2000). Используя мышей и крыс, было установлено, что pendrin экспрессируется на апикальных частях мембран тироидных, почечных и внутреннего уха клетках. Отсутствие pendrin, как полагают, непосредственно отвечает за дефекты органофикации iodide у Pendred пациентов. Однако, Slc26a4 нокаутные мыши лишены тироидных симптомов (Everett et al., 2001) , а точная роль pendrin в щитовидной железе всё ещё неясна. Во внутреннем ухе мышей pendrin обнаруживается в апикальных частях мембран клеток, покрывающих эндолимфатические полости, это расценивается как участие в гомеостазе эндолимфы (Everett et al. , 1999; Royaux et al., 2003; Yoshino et al., 2004). Кроме того, экспрессия pendrin в улитке включает также поддерживающие клетки кортиева органа (Claudius and Deiters' cells), также как и спиральную лигаменту и спиральный ганглий. Недавно более чувствительный подход (postembedding immunogold анализ с помощью ЭМ) выявил некоторую экспрессию pendrin также в OHC
и IHC, особенно в их апикальных частях мембран и стереоцилиях (Yoshino et al., 2006).
Slc26a4
-/- нокаутные мыши (Pds) обнаруживают waltzer-подобную вестибулярную дисфункцию и полную глухоту. Внутреннее ухо у них развивается нормально вплоть до ст.l E15,два дня спустя после начала экспрессии pendrin у мышей дикого типа. Следовательно, тяжелая дилятация эндолимфатической полости развивается как в улитке, так и преддверии (Figure 2, K-L). Это расширение, как полагают, является вторичным по отношению измененным осмотическим условиям и увеличенному объему эндолимфатической жидкости. Во время второй постнатальной недели волосковые клетки начинают дегенерировать В преддверии отолиты и отолитовые мембраны также деструктивны (Everett et al., 2001). После отнятия от груди маргинальные клетки strial vascularis Pds
-/- мышей обнаруживают нерегулярную форму и размер, что ведет в результате к истончению stria vascularis. У взрослых Pds
-/- мышей гиперпигментация клеток strial vascularis предшествует их дегенерации, указывая на повреждения свободными радикалами. Функциональные эксперименты выявили, что Pds
-/- мыши постепенно теряют EP, начиная с P12, ещё до начала нормального слуха. Тем не менее эндолимфатическа концентрация K
+ и экспрессия Kcnq1/Kcne1 каналов были нормальными. Дефицит pendrin устраняет также экспрессии Kcnj10 K
+ каналов в strial intermediate клетках, хотя мРНК Kcnj10 экспрессируется нормально (Royaux et al., 2003; Wangemann et al., 2004). Kcnj10 каналы выполняют роль в рециклинге ионов K
+ поперек барьера базальных клеток stria vascularis. Т.о., pendrin может выполнять роль по поддержанию EP не затрагивая секрецию K
+ маргинальными клетками stria vascularis, но скорее всего влияет на токи K
+ в промежуточных клетках. Kcnj10 нокаутные мыши не генерируют EP, но обладают пониженными эндолимфатическим объемом и концентрацией K
+ (Marcus et al., 2002). Следовательно, дефицит pendrin может давать дополнительные эффекты. Др. роль pendrin была недавно выявлена в улитке (Wangemann et al., 2007) и преддверии (Nakaya et al., 2007). Ca
2+ каналы (Trpv5 и Trpv6) в эпителиальных клетках преддверия и улитки резорбируют ионы кальция из эндолимфы и ингибируются с помощью низкого pH. В улитке Trpv5 и Trpv6 экспрессируются в маргинальных клетках strial vascularis и эпителиальных клетках бороздки, соотв. Эти каналы поддерживают низкую концентрацию Ca
2+ нормальной эндолимфы. Pendrin-нокаутные мыши обнаруживают более низкий pH и более высокую концентрацию Ca
2+ в эндолимфе, приводя к снижению трансэпителиального потенциала в utricle. Более высокий уровень Ca
2+ в эндолимфе может ингибировать сенсорную трансдукцию, необходимую для слуха и способствует дегенерации волосковых клеток. Т.о., во внутреннем ухе pendrin, как полагают, функционирует как Cl
-/HCO
3- , который обеспечивает секрецию щелочных HCO
3- ионов в эндолимфатическое пространство и одной из важнейших его ролей может быть поддержание эндолимфатического pH (Nakaya et al., 2007; Wangemann et al., 2007). Гиперпигментация stria vascularis у взрослых Pds
-/- мышей делает возможным предположение, что воспалительный процесс участвует в их дегенерации. В самом деле, гиперпигментация и реорганизация маргинальных клеток возникают конкурентно с инвазией макрофагов и complement маркеров (Jabba et al., 2006). Предполагается, что winged helix/forkhead ген
Foxi1 (известный также как Fkh10 ) индуцирует экспрессию pendrin , т.к. Foxi1-нулевые мыши не экспрессируют pendrin и обнаруживают фенотип, сходный с таковым у pendrin нокаутных мышей (Hulander et al., 2003).
SLC26A5/prestin - the motor protein of outer hair cell
electromotility
В улитке млекопитающих представлены два механизма умножения звуковых сигналов: (a) умножение движения стереоцилий с помощью механо-электрических каналов трансдукторов (существует во всех известных слуховых органах); и (b) OHC somatic electromotility - voltage-зависимое быстрое изменение длины и жесткости OHC (существует только во внутреннем ухе млекопитающих), называемое также улиточный умножитель (cochlear amplifier). Электроподвижность включает укорочение деполяризованных OHC и удлинение гиперполяризованных клеток, независимо от уровней АТФ или OHC Ca2+. Умножение с помощью OHC электроподвижности, как полагают, умножает вибрации в улитке и делает возможным более острый слух и частотную избирательность в улитке млекопитающих. Этот механизм позволяет отвечать улитке на низко (мене 1 KHz) частотные сигналы [rev. (Frolenkov, 2006)].
Prestin является интегральным белком, который экспрессируется только в OHC улитки (Figure 1D). Молекулы Prestin, как мономеры, так и тетрамеры, обильно экспрессируются вдоль латеральной части мембраны OHC и в меньшей степени - в базальной части мембраны. Онтогенетическая экспрессия
prestin совпадает с появлением электроподвижности (Belyantseva et al., 2000; Zheng et al., 2000; Yu et al., 2006). Хотя prestin относится к семейству SLC26 обменников анионов и имеет сходную структуру с др. членами этого семейства, четкие доказательства демонстрируют, что он действует как ещё не описанный транспортер ионов. Более того, нокаут prestin (Slc26a5) у мышей не затрагивает токов во всей OHC клетке (Liberman et al., 2002). Вместо этого, prestin рассматривается как voltage-зависимый моторный белок, ответственный за электроподвижность OHC (Zheng et al., 2000), т.к. Slc26a5 нокаутные мыши не обнаруживают OHC electromotility (Figure 2M) и избирательности частот. Эти мыши подтверждают предположение, что электромобильность OHC усиливает чувствительность внутреннего уха, т.к. они обладают потерей чувствительности улитки к 40-60 dB без нарушения пучков волосков OHC и механо-электрической передачи. Кроме того, Slc26a5-нулевые мыши обладают более короткими OHC (Figure 2N), что не удивительно, т.к. prestin очень обилен в латеральных стенках этих клеток. В возрасте 4-9 недель обнаруживается вторичный апоптоз OHC в базальной четверти улитки у Slc26a5-нулевых мышей, сопровождаемый дегенерацией IHC, хотя IHC не экспрессируют prestin. Однако, HI предшествует дегенерации волосковых клеток, по крайней мере, за две недели, указывая тем самым, что электроподвижность является первичной причинойпотери слуха (Liberman et al., 2002; Cheatham et al., 2004; Wu et al., 2004). Недавно типичное распределение prestin вдоль латеральной части мембраны OHC обнаружило зависимость от рецептора тироидного гормона TR (Winter et al., 2006). Хотя абсолютная величина электромобильности OHC у гетерозиготных мышей составляла половину от нормы (Liberman et al., 2002), функция и вид улитки у мышей без одной копии Slc26a5 были нормальными (Cheatham et al., 2005). Было подтверждено, что prestin ощущает напряжение (voltage) благодаря связыванию внутриклеточных ионов Cl
- в деполяризованных клетках. Как результат его конформация изменяется. Т.о., prestin является очень эффективным прямым voltage-to-force конвентором. Его функция ассоциирована с типичным нелинейным ёмкостным сопротивлением, которое измерено [rev. (Dallos et al., 2006)].
Unconventional myosins
Нестандартные миозины являются моторными молекулами. которые содержат актин-связывающий домен на своём N-терминальном моторном или головном домене. Используя АТФ в качестве энергетического источника они могут двигаться вдоль актиновых филамент. Нестандартные миозины обладают также сайтами связывания для белков на своем C-терминальном хвосте и тем самым они могут служить в качестве "cars", которые перемещают белки грузы к их местам целям в клетке. Внутреннее ухо млекопитающих экспрессирует несколько неконвенционных миозинов, каждый из которых имеет уникальный паттерн экспрессии и функции во внутреннем ухе. Мутации в 5 генах миозинов (Myo1a, Myo3a, Myo6, Myo7a и Myo15a) ассоциированы с HHL у людей. Паттерн экспрессии myosin 1A во внутреннем ухе мыши ещё не изучен. Myo3a, Myo6, Myo7a и Myo15a экспрессируются в волосковых клетках внутреннего уха мышей и выполняют роль в организации пучков волосков [rev. (Hertzano and Avraham, 2005)]. Два из них, myosins VIIa и XV, м. связывать PDZ сайты на harmonin или whirlin и являются частью Usher-связанной сети, которая представлена на Figure 1F [rev. (Reiners et al., 2006)]. Т.о., myosin VIIa (Boeda et al., 2002; Senften et al., 2006) и myosin XVa (Belyantseva et al., 2005) активно транспортируют harmonin и whirlin, вместе с прикрепленными к ним белками в соотв. места стереоцилий. Недавно было показано. что myosin IIIa также локализуется на кончикахх стереоцилий и необходим для собственно их поддержания (Schneider et al., 2006). Существуют мышиные модели только для мутаций Myo6, Myo7a и Myo15a. Нулевые мутации Myo6 [Snell's waltzer (Avraham et al., 1995)], Myo7a [shaker1 (Self et al., 1998)] и Myo15a [shaker2 (Probst et al., 1998)] дают сходные waltzer-подобные фенотипы у гомозигот (глухота и вестибулярная дисфункция), в результате слияния стереоцилий ( Myo6), дезорганизации (Myo7a) или укорочения (Myo15a). Myo6 и Myo7a нулевые мыши обнаруживают также последующую дегенерацию волосковых клеток. В то время как мыши, гомозиготные по нулеваым мутациям Myo6, Myo7a или Myo15a глухи, гетерозиготы имеют нормальный фенотип. Более того, двойные гетерозиготные мыши по Myo15a и др. (Myo6 or Myo7a) нулевому аллелю также были нормальными (Karolyi et al., 2003). Однако, missense Myo7a мутация (headbanger mice; Hdb) индуцируют вестибулярный фенотип и умеренную HI также у гетерозигот
(Figure 3D), в результате элонгации и слияния стереоцилий волосковых клеток. Гомозиготы имеют более тяжелый фенотип (Rhodes et al., 2004). Myo6 проанализирован более детально.
Спонтанная мутация Snell's waltzer (sv), возникла в 1966 (Deol and Green, 1966). Поведение кружения Snell's waltzer мышей представлено на Figure 3E. Индуцированная облучением мутация в том же локусе также доступна (se
sv/se
sv) (Russell, 1971), как и индуцированная ENU мутация (ENU89) (personal communcation, Colin Fletcher and Karen Avraham). Мутация в гене Myo6 была найдена в
sv аллеле (Avraham et al., 1995). Во внутреннем ухе мышей и рыбок данио myosin VI экспрессируется специфически в апикальной плазматической мембране волосковых клеток вблизи основания стереоцилий (Self et al., 1999; Kappler et al., 2004). Гомозиготные
sv мыши обнаруживают прогрессирующу дегенерацию волосковых клеток внутреннего уха со ст. P12, приводя к дегенерации всего нейроэпителия внутреннего уха. Ранние стадии развития волосковых клеток и стереоцилий протекают нормально,так что при рождении дезорганизована только часть пучков волосков. Однако, во время первой постнатальной недели дезорганизуются пучки волосков прогрессивно и апикальная плазматическая мембрана волосковых клеток. Затем в течение слудующих 2-х недель стереоцилии аномально сливаются др. с др., чтобы сформировать гигантские не функциональные стереоцилии (Self et al., 1999; Kappler et al., 2004). Сходный фенотип наблюдается у рабок данио с мутацией гена
Myo6b . У них ген
Myo6 удвоен (
Myo6a и Myo6b) и только
Myo6b обнаруживает предпочтительную экспрессию во внутреннем ухе и нейроэпителии латеральной линии. Подобно
sv мышам мутации у рыбок данио
Myo6b ответственны за слуховые и вестибулярные дефекты (мутантны
satellite) из-за дезорганизации пучков волосков, в которых стереоцилии в конечном счете сливаются. Структурные дефекты апикальной плазматической мембраны, проявляющиеся в виде крупных пузырьков, накапливаются вблизи кутикулярной пластинки (Seiler et al., 2004). Базируясь на мутантах
satellite у рыб и
sv мышей, было предположено, что myosin VI закрепляет апикальную плазматическую мембрану стереоцилий на стержневых актиновых филаментах (Figure 1F). В отсутствие myosin VI апикальная плазматическая мембрана выпячивается выше эпителия и между стереоцилиями, что и ведет к слиянию стереоцилий. Мутации в гене
MYO6 человека сцеплены с HHL. В то время как у мышей мутации
Myo6 ассоциируют только с рецессивными NSHL, мутации
MYO6 у людей были сцеплены как с доминантными
(Melchionda et al., 2001) , так и рецессивными NSHL (Ahmed et al., 2003), а также с доминантными SHL, которые включают кардиальную гипертрофию и удлинение QT в дополнение к сенсоронейральной HHL (Mohiddin et al., 2004).
Hair cell genes for transcription factors
Сенсорные волосковые клетки и не-сенсорные поддерживающие клетки в нейроэпителии внутреннего уха возникают из общих предшественников.
TABLE 2
GENES THAT WERE LINKED WITH HUMAN HHL AND CLONED, BUT HAVE NO MUTANT MOUSE MODEL THUS FAR
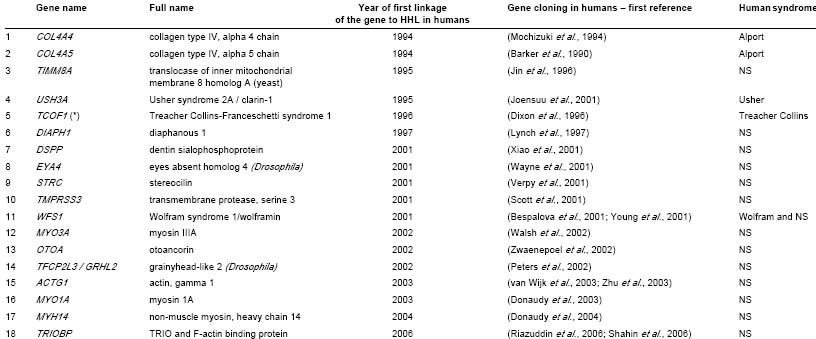
NS, only non syndromic hearing loss; (*) There is a Tcof1-knockout mouse model. Heterozygous mice exhibited severe craniofacial malformations, including malformations of external and internal
ear, and died at birth (Dixon et al., 2000; Dixon et al., 2006). However, the effect Tcof1 haploinsufficiency on the mice inner ears has not been reported.
Просенсорные клетки предшественники дифференцируются в волосковые клетки в обязательном порядке (default) , но это решение о дифференцировке обычно ингибируется передачей сигналов Notch
(Yamamoto et al., 2006). Активация Notch латерально репрессирую экспрессию
Math1/Atoh1 транскрипционного фактора, что необходимо вместе с Sox2 (described below), чтобы индуцировать дифференцировку просенсорных клеток предшественников в волосковые клетки. В самом деле, нокаутные
Atoh1 мыши не имеют волосковых клеток в преддверии и улитке (Bermingham et al., 1999; Yamamoto et al., 2006).
Многие дополнительные транскрипционные факторы являются критическими для развития внутреннего уха. Некоторые из них экспрессируются специфически в волосковых клетках внутреннего уха (e.g. Pou4f3) , а др. являются критическими для развития др. органов также. Некоторые транскрипционные факторы коррелируют с глухотой как у людей, так и мышей ( Eya1, Pou3f4, Pou4f3, Mitf, Pax3, Snai2/Slug, Sox2, Sox10, Six1 ; see Supplementary Table S1). Sox2 и Pou4f3 будут описаны в качестве примера.
SOX2 мутации у людей коррелируют в основном с двухсторонней anophthalmia (глазными аномалиями) у гетерозигот (Fantes et al. , 2003). Однако, две de novo SOX2 мутации коррелировали с SHL у гетерозигот. Nonsense мутация (Q155X),как полагают, ответственна за HI, в дополнение к анофталмии, отсутствию всех оптических путей и к др. нейрологическим аномалиям (Hagstrom et al., 2005); а missenseмутация (479delA) , как было подтверждено, ответственна за синдром комбинированного врожденного hypothalamo-pituitary нарушения и HI (Kelberman
et al., 2006). У эмбрионов мышей Sox2 экспрессируется в основном в развивающейся ЦНС и сенсорных плакодах, где он играет критические роли в эмбриогенезе. На ст. E9.5 Sox2 экспрессируется не только в нервной трубке. но также в отоцисте, из которого и развивается нейроэпителий внутреннего уха. В развивающейся улитке Sox2 обычно экспрессируется только в клетках просенсорных предшественников, также как и в дифференцированных волосковых и поддерживающих клетках развивающегося кортиева органа (Wood and Episkopou, 1999; Kiernan et al., 2005). Два мутантных Sox2 аллеля, Lcc (light coat and circling) и Ysb (yellow submarine), были получены у мышей. Мыши, несущие мутантные аллели могут быть легко идентифицированы из-за полудоминантной желтой окраски шерсти. Аллели Lcc и Ysb содержат интактные кодирующие и соседние последовательности к Sox2, но регуляторные элементы, которые затрагивают экспрессию Sox2 мутантны (вставленная последовательность, использованная для получения аллеля Ysb, содержит регуляторную последовательность от гена Col2a1). Как результат, Lcc и Ysb гомозиготы E9.5 эмбрионов мышей экспрессируют нормальный Sox2 в нервной трубке, но нет (Lcc) или мало (Ysb) Sox2 в отоцисте. Т.о., мутации не нарушают развития головного мозга, индуцируя умеренные фенотипы по сравнению с SOX2 мутациями, которые описаны у людей. Гомозиготные мыши обладают тяжелыми HI (Ysb мыши) или полной глухотой (Lcc мыши), а также поведнием кружения, из-за нарушений внутреннего уха и его нейроэпителия. Преддверие затронуто более тяжело. При рождении Lcc мыши, которые не экспрессировали Sox2 во внутреннем ухе обнаруживали более тяжело изуродованные внутренние уши, а нейроэпителий отсутствовал полностью, поэтому ни волосковые, ни поддерживающие клетки не дифференцировались. Ysb гомозиготы, которые экспрессировали на низком уровне Sox2 во внутреннем ухе, почти не обнаруживали волосковых клеток в свих преддвериях. В базальной части улитки Ysb гомозиготы обнаруживали аномальные участки дезорганизованных волосковых клеток, с регионами, не содержащими волосковых клеток между ними (Figure 2, G-J). Апикальная область улитки включала дезорганизованные волосковые клетки с отсутствием четкого выравнивания IHC и OHC. Уникальный паттерн развития волосковых клеток у Ysb мышей может быть результатом вставленного регуляторного Col2a1 мотива в регуляторные последовательности Sox2. Lcc гомозиготы, которые не обнаруживают экспрессии Sox2 во внутреннем ухе, не экспрессируют Atoh1 , тогда как Ysb гомозиготы, которые экспрессируют Sox2 экспрессируют и Atoh1. Следовательно, Sox2 , как полагают действует выше Atoh1 (Dong et al., 2002; Kiernan et al., 2005).
Гены POU-доменовых транскрипционных факторов, как известно, контролируют терминальные стадии развития ЦНС [rev. (Ryan and Rosenfeld, 1997)]. У мышей
Pou4f3 (известен также как Brn-3c или Brn3.1) экспрессируется очень специфически в волосковых клетках улитки (Figure 2O) и преддверия. Его экспрессия может быть установлена в волосковых клетках внутреннего уха со ст. E12.5, после экспрессии
Atoh1 и она постепенно усиливается вплоть до рождения (Xiang et al.,
1998; Hertzano et al., 2004). Мутация
POU4F3 сцеплена с аутосомно доминантной прогрессивной NSHL у людей (Vahava et al. , 1998). Pou4f3-нокаутные мыши (Erkman et al., 1996; Xiang et al. , 1997), также как и dreidel (
ddl) мыши, которые не экспрессируют функционального Pou4f3 (Hertzano et al., 2004), обнаруживают сходный waltzer-подобный фенотип с выраженной глухотой (Figure 3, A-B) и вестибулярную дисфункцию, включая трясение головы, поведение кружения и гиперактивность. Pou4f3-нокаутные мыши обнаруживают прогрессирующую потерю волосковых клеток внутреннего уха в улитке и преддверии, что ведет ко вторичной дегенерации поддерживающих клеток, а также к дегенерации нейронов спирального и вестибулярного ганглиев. Pou4f3 экспрессируется в постмитотических просенсорных клетках предшественниках, которые детерминированы к развитию в волосковые клетки, но не в pre-commitment митотических клетках. Волосковые клетки в развивающемся внутреннем ухе Pou4f3 нокаутных мышей подвергаются инициальной дифференцировке, но не способны формировать зрелые стереоцилии, некоторые волосковые клетки оказывались неправильно локализованы в слое поддерживающих клеток и все или большинство из них дегенерировали вследствие апоптоза во время беременности и первые дни после рождения. Т.о., Pou4f3 является критическим для нормальной терминальной дифференцировки, миграции и жизнеспособности волосековых клеток внутреннего уха (Erkman et al., 1996; Xiang et al. , 1997; Xiang et al., 1998). Гены транскрипционных факторов Gfi1 и Lhx3 предполагаются в качестве мишеней для Pou4f3. Гомозиготные
dreidel мыши экспрессируют минимальные уровни мРНК Gfi1 в волосковых клетках улитки и преддверия (Figure 2P) и не экспрессируют Lhx3 в волосковых клетках улитки (Figure 2, S-V) (Hertzano et al., 2007).
Deaf mouse mutants not correlated with human heredi-
tary hearing loss
Мутантные мышиные модели еще не получены для всех генов, которые сцеплены с HHL у людей. Из 61 клонированного гена, которые ассоциируют с HHL у людей (Van Camp and Smith, 2006), по 18 не получены модельные мыши (Table 2). 75% генов, которые были сцеплены с уродствами или с дисфункцией во внутреннем у мышей (Jackson_Laboratory, 2007) не были ещё сцеплены с HHL у людей (Van Camp and Smith, 2006). Продукты некоторых из этих генов могут взаимодействовать с уже известными связанными с глухотой сетями. Напр., лизосомный мембранный белок Scarb2/LIMP-2, который регулирует мембранный транспорт некоторых белков, как было установлено, важен для локализации Kcnq1/Kcne1 калиевых каналов в апикальной мембране маргинальных клеток stria vascularis и в вестибулярных темных клетках взрослых мышей. Как результат Scarb2-дефицитные мыши обладают прогрессирующей потерей слуха из-за дегенерации stria vascularis (Knipper et al., 2006).
Др. гены, которые были сцеплены с HI у нокаутных мышей представляют класс продуктов, которые ещё не связаны с HHL у людей, но, по-видимому, являются критическими для функции внутреннего уха. Напр., creatine kinase (Ckb) может играть роль в переносе АТФ на ATPases стереоцилий.
Ckb нокаутные мыши обнаруживают HI и вестибулярную дисфункцию. Цитозольная изоформа из головного мозга creatine kinase является наиболее многочисленным белком после β-actin в пучках волосков utricle птиц (Shin et al., 2007).
Др. недавним примером является спонтанная мутация (
jbg) , которая ведет к HI и вестибулярной дисфункции у мышей
jitterbug , она картирована в гене
Clic5. Clic5 принадлежит к семейству хлорных внутриклеточных каналов. Во внутреннем ухе мышей он специфически выявляется в базальной части стереоцилий волосковых клеток улитки и преддверия.
Jitterbug мыши обнаруживают аберрантные стереоцилии и прогрессирующую дегенерацию волосковых клеток, это указывает на то, что Clic5 может играть роль в сборке или поддержании стереоцилий. Clic5, по-видимому, ассоциирует с radixin в основании стереоцилий и как полагают. участвует в формировании или стабилизации соединений между плазматической мембраной стереоцилий и их актиновой сердцевиной (Gagnon et al., 2006). Та же самая мутация может индуцировать разные фенотипы в разных инбредных линиях мышей, которые экспрессирую разные генетические модификаторы, такие модификаторы были также описаны у людей [первым был локус DFNM1 (Riazuddin et al., 2000)]. У мышей этот феномен известен как эффект линейного фона. Напр.,
mdfw и
Ahl аллели гена
Cdh23 могут индуцировать возраст-зависимую и индуцируемую шумами потерю слуха у гомозигот нескольких линий мышей (с разным временем начала в разных линиях), но др. линии оказались относительно резистентны к этим мутациям (Zheng and Johnson, 2001; Noben-Trauth et al., 2003). Мутантные аллели
Cdh23 могут действовать как генетические модификаторы у гетерозигот. Т.о., аллель
Ahl может модифицировать потерю слуха у Mass1
frings мутантных мышей (Johnson et al., 2005). По крайней мере, дополнительно 7 локусов могут индуцировать связанную с возрастом потерю слуха у мышей. У двугенных мутантных мышей, которые несут два мутантных гена и обладают др. фенотипом по сравнению с мышами, гомозиготными по мутации одного из генов, могут рассматриваться как генетические модификаторы [e.g. (Adato et al., 1999; Johnson et al., 2005; Zheng et al., 2005)]. Чтобы идентифицировать такие модификаторы, некоторые группы скрещивали мышей, несущих мутации, связанные с глухотой с мышами различных линий [e.g. (Asher et al., 1996; Niu et al., 2006; Johnson
et al., 2006)].
Summary
Efforts are now underway to create knock-outs and conditional
mutants for every gene in the mouse genome [NIH knockout
mouse project, (KOMP): http://www.nih.gov/science/models/
mouse/knockout/; and European conditional mouse mutagenesis
program (EUCOMM): http://www.eucomm.org/]. This endeavor
will undoubtedly create many more mouse models for human
HHL. As discussed above, there have been many cases where
the mouse gene has led to the discovery of the human HI gene
(and vice versa), emphasizing the complementarity of mouse and
human studies in the auditory and vestibular systems. Complex
hearing impairment, which includes both noise-induced hearing
loss and age-related hearing loss (presbyacusis), as well as the
identification of modifiers, will require additional mouse models.
The identity of a human mutation is critical for human diagnostics
and genetic counseling, and early identification and intervention
is beneficial for hearing impaired patients (White, 2004; Hyde,
2005). The information acquired from mouse morphological and
physiological studies, as exemplified from the various techniques
in Figure 2, demonstrates that the study of mouse models for
deafness will undoubtedly provide a key to understand auditory
function and help develop critical elements for therapeutics (Atar
and Avraham, 2005; Tang et al., 2006a).
Сайт создан в системе
uCoz