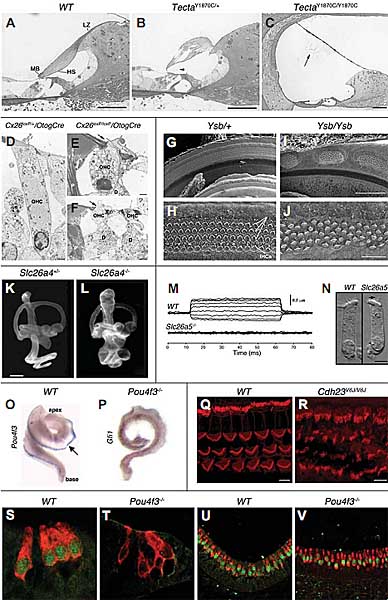
Length changes of OHC in response to voltage steps (-120-60 mV in 20 mV steps) in whole-cell, voltage-clamp recordings. (N) Micrographs of OHC isolated from apical turns of cochleae. OHC that do not express prestin are shorter than wild type OHC and do not exhibit electromotility. Arrows in (N): open arrow - nucleus; filled arrow - stereocilia. Scale bar, 5 µm in (N). Reprinted with permission from (Liberman et al., 2002). (O-P) Whole mount in situ hybridizations (ISH) detect expression of Pou4f3 (O) and Gfi1 (P) mRNAs in E18.5 cochleae from wild type (O) and dreidel (P) littermate mice. Dreidel mice, which do not express functional Pou4f3 protein, do not express Gfi1 mRNA. Arrow - Pou4f3
mRNA expression is detected as a blue band along the lateral wall of the cochlea. Reprinted with permission from (Hertzano et al., 2004). (Q-R) Whole mount immunohistochemistry detects spatial expression pattern of F-actin (shown in red, stained with rhodamine phalloidin) in stereocilia in the middle turn of wild type (Q) and waltzer V 6J (R) organ of Corti at P7. The V 6J allele was reported to be a functional null allele of Cdh23 (Di Palma et al., 2001a; Di Palma et al., 2001b). Waltzer mice exhibit disorganized stereocilia (Lagziel et al., 2005). Scale bars, 5 µm. Figures from Ayala Lagziel and Thomas B. Friedman. (S-V) Immunohistochemistry of paraffin sections of E18.5 wild-type (S and U) and Pou4f3-/- (T and V) mouse inner ears. Expression of Lhx3 (green) and myosin VI (red) was detected in the cochlea (S-T) and the vestibular system utricle (U-V). Whereas Lhx3 is expressed in the nuclei of all hair cells in the wild-type inner ears, Lhx3 expression could be detected only in the vestibular system of the Pou4f3-/- mice but not in any of the nuclei of the cochlear hair cells (Hertzano et al., 2007). Figures from Amiel Dror.
Extracellular matrix components: cartilage and tectorial membrane defects (collagen genes and Tecta)
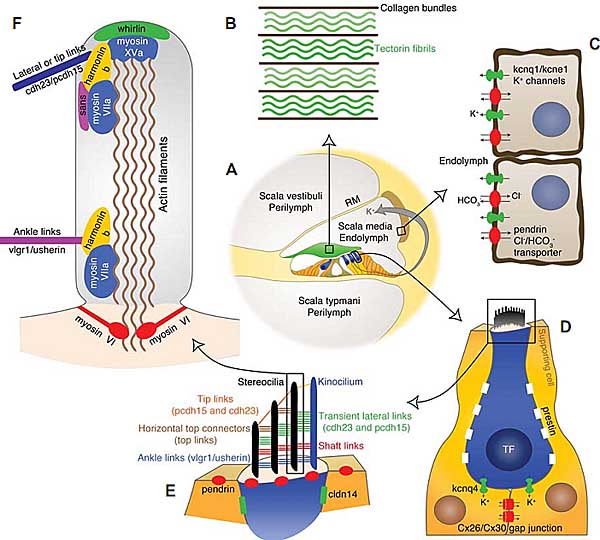
Орган слуха млекопитающих, орган Корти, находится в улитко-подобном образовании cochlea на полосе соединительной ткани, the basilar membrane (BM). Базирующаяся на коллагене BM дифференцирована по жесткости вдоль улитки и вибрирует в ответ на вызываемые звуками движения кохлеарной жидкости. Эти вибрации воспринимаются двумя типами волосковых клеток, включенными в сенсорнный эпителий Кортиева органа, внутренними и наружными волосковыми клетками (IHC и OHC, соотв.). Механосенсорные пучки волосков из OHC проецируются вверх из ретикулярной ламины, апикальной поверхности сенсорного эпителия и внедряются в лежащую поверх текториальную мембрану (TM) [rev. (Raphael and Altschuler, 2003)]. Поперечный разрез кортиевого органа проиллюстрирован на Рис. 1A. TM млекопитающих имеет уникальную и высоко организованную ультраструктуру. Она содержит две основные группы компонентов: коллагеновые фибриллы, которые организованы в тяжелых пучков и идут радиально через TM, и гликопротеинов, которые состоят из необычайно исчерченного слоя матрикса, окружающего фибриллы (Figure 1B) (Hasko and Richardson, 1988). Коллагены типов II, IX и XI составляют радиальные фибриллы(Slepecky et al., 1992; Thalmann, 1993), в то время как два гликопротеина, alpha и beta tectorins (кодируемые Tecta and Tectb), являются основными компонентами матрикса TM (Legan et al., 1997). 7 коллагеновых белков оказались связанными в HHL человека: COL2A1, COL4A3, COL4A4, COL4A5, COL9A1, COL11A1 и COL11A2 (Van Camp and Smith, 2006). Только 5 из них обнаруживаются у модельных мышей (Supplementary Table S1). Мутация в COL11A2 сцеплена с аутосомно доминантной NSHL у людей ( DFNA13 locus), а также с синдромом Stickler. Др. коллагеновые гены были сцеплены только с SHL у людей, в основном с Stickler ( COL2A1,
COL9A1 и COL11A1) и Alport (COL4A3-5) синдромами. Связанные с Alport синдромом коллагены (цепочки alpha-3, 4 и 5 коллагена типа IV) включены в базальные мембраны внутреннего уха и почечных клубочков. В улитке они экспрессируется BM, частях спиральной лигаменты и в stria vascularis. Как результат синдром Alport (Alport, 1927) характеризуется нейросенсорной HHL и прогрессирующим нефритом, часто приводящим к почечной недостаточности [rev. (Hudson et al., 2003)]. После идентификации мутаций в гене COL4A3 человека, как ответственных за синдром Alport (Mochizuki et al., 1994), Col4a3 был нокаутирован у мышей (Cosgrove et al., 1996). Гомозиготы погибали в возрасте 14 недель из-за почечной недостаточности. Дефекты базальных мембран обнаруживались в почечных клубочках и мембранозном лабиринте улитки, как и при болезни у людей. Почечный фенотип включает прогрессирующий гломерулонефрит с протеинурией и микрогематурией, с локальными мультислойными утолщениями и истончениями базальных мембран гломерул, а также с фибротическими гломерулами со спавшимися капиллярами. В мембранозном лабиринте улитки обе цепочки Col4a3 и Col4a4 полностью отсутствуют. Базальные мембраны специфических частей мембранозного лабиринта были очень тонкими, толстыми или не обнаруживались в сравнении с улитками дикого типа, а соседние капилляры были спавшимися. И почечные и кохлеарные дефекты прогрессировали и HI обнаруживалась только после доститжения возраста 6 недель (Cosgrove et al. , 1996; Cosgrove et al., 1998). Stickler синдром (Stickler et al., 1965) включает помимо прогрессирующей нейросенсорной HHL, преждевременные дегенеративные изменения в различных суставах с аномальным развитием эпифизов, аомалиями позвонков, остеоартритами и иногда также с необычным лицом и расщеплением нёба. Известны три типа синдрома Stickler: type 1 включает также прогрессирующую миопатию и слепоту из-за vitreoretinal дегенерации и отслойки сетчатки, тогда как тип 2 обладает др. дефектами стекловидного тела без отслойки сетчатки [rev. (Snead and Yates, 1999)]. Type 3 более умеренный без миопатии и глазных нарушений (Vikkula et al., 1995). Связанные с синдромом Stickler коллагены Col2a1, Col11a1 и Col11a2 являются важными компонентами не только кохлеарной TM, но и также хрящей (Col2a1 экспрессируется также в стекловидном теле глаз). Т.к. внутреннее ухо содержит хрящевое покрытие, которое играет важную роль в его эмбриогенезе, то мутантные коллагены типов II и XI влияют на размеры, структуру и развитие внутреннего уха.
COL2A1 участвует в нейросенсорной глухоте, которая сопровождается несколькими сходными наследственными синдромами у людей, такими как Stickler syndrome, spondyloepiphyseal dysplasia congenita (SEDC) и chondrodysplasia. Dmm (autosomal semi-dominant disproportionate micromelia), мыши с мутантным геном Col2a1 получены в 1966 от потомства самцов с облученными сперматогониями. Мутация Dmm является делецией 3-х нуклеотидов в области, кодирующей C-propeptide глобулярный домен Col2a1. Делеция ведет к замещению двух аминокислот, Lys иThr, на одиночную аминокислоту, Asn, в мутантном белке (Pace et al., 1997). Dmm экспрессируют пониженные уровни коллагена II и страдают от дефектов хрящей, которые затрагивают развитие внутреннего уха также. Гомозиготы являются карликами с диспропорционально коротки конечностями (micromelia), имеют расщепление нёба (Brown et al., 1981; Seegmiller et al., 1988) и погибают при рождении из-з легочной гипоплазии (Foster et al., 1994). Внутреннее ухо гомозиготных Dmm эмбрионов содержит меньше коллагеновых волокон и характеризуется нерегулярной цитодифференцировкой хондроцитов во внеклеточном матрикса по сравнению с эмбрионами дикого типа (Berggren et al., 1997). Как результат дисморфогенез отической капсулы и перилимфатических пространств во время эмбриогенеза, это ведт к развитию уродливого внутреннего уха с объёмистой хрящевой капсулой и отсутствием или уменьшением определённых перилимфатических пространств (Van de Water and Galinovic-Schwartz, 1987). Недавно спонтанно возникла missense мутация гена Col2a1 (R1417C) у мышей. Эти мыши были названы sedc, т.к. их фенотип был сходен с spondyloepiphyseal dysplasia congenita человека. Гомозиготные sedc взрослые мыши имеют укороченный нос, диспластичные позвоночник, бедра и большеберцовые кости, retinoschisis и потерю слуха (Donahue et al., 2003). Мутагенез с помощью генного таргетинга использован для получения Col2a1 G574S мышей, которые служат моделью хондродисплазии, т.к. обнаруживают параллелизм с мутацией, обнаруженной у людей. В дополнение к скелетным аномалиям мыши обнаруживали нарушения слуха из-за образования неправильной формы отической капсулы. В то время как нормальная отическая капсула округла, трансгенная отическая капсула уплощена и удлинена. Предполагается, что более слабый хрящ отической капсулы не может противостоять механическому давлению от развивающихся лица и головного мозга и расплющивается (Maddox et al., 1998). Гетерозиготные Col2a1 мутантные мыши обладают более умеренным, но не нормальным фенотипом.
Cho мыши появились спонтанно в 1971 (Seegmiller et al., 1971). Гомозиготы имеют расщепление нёба и погибают вскоре после рождения из-за летальной хондродисплазии. Мутация cho является делецией 1-nt в гене Col11a1, это вызывает сдвиг рамки считывания и возникновение преждевременного кодона терминации, приводя к образованию укороченного генного продукта, который не может формировать ансамбли с др. молекулами коллагена. Т.о., cho действительно является функциональным нулевым аллелем Col11a1 (Li et al., 1995). HГомозиготы имеют тяжелые нарушения слуха при рождении из-за недоразвития Кортиева органа в нижнем витке улитки, с отсутствием волосковых клеток, поддерживающих клеток, нервных окончаний и pillar клеток (Cho et al., 1991). Т.к. гетерозиготные cho (аллель Col11a1) страдают от возараст-зависимых остеоартритов, было предположено, что аллель cho может оказывать деструктивный эффект на соединительную ткань. Однако гетерозиготные мыши хорошо слышат во время первых двух месяцев жизни, а умеренная или прогрессирующая потеря слуха у них развивается позднее (обусловленность возрастом),, она не значительно отличается от мышей дикого типа (Szymko-Bennett et al., 2003). В противоположность мышам человеческая COL11A1-сцепленная SHL проявляется также у гетерозигот: точковая мутация в COL11A1 (G97V) оказалась сцепленной с аутосомно доминантным синдромом Stickler (Richards et al., 1996) , а мутация сплай-донорского сайта в этом гене оказалась связанной со сходным аутосомно доминантным Marshall синдромом (Griffith et al., 1998).
Две мышиные модели с целенаправленно полученными мутациями в Tecta (alpha tectorin) были получены той же самой группой. Обе мут ации индуцируют дефекты TM и HI. Первой мутацией была целенаправленная делеция в Tecta (наз. Tecta
Δ ENT). Единственный дефект у гомозиготных мышей, которые не экспрессируют alpha tectorin (нулевая мутация), наблюдается в TM, которая лишена всего не-коллагенового матрикса и полностью отсоединена от Кортиева органа и спирального. Внутреннее ухо у них было менее чувствительным к звуковым стимулам, это подкрепляет гипотезу, что TM умножает реакцию волосяных клеток в ответ на низкие уровни сигналов (Legan et al., 2000; Lukashkin et al., 2004). Изучение гомозиготных Tecta
ΔENT
мышей вместе с изучением подвижности TM и BM Кортиева органа [e.g. (Hemmert et al., 2000)], помогло выяснению роли этих мембран [For details, see (Legan et al. , 2005)]. Вторая мышиная модель, несущая missense мутацию в Tecta (Legan et al., 2005), идентичную мутации Y1870C, которая была обнаружена у людей с нарушениями слуха (Verhoeven et al., 1998). Гомозиготные Tecta Y1870C/Y1870C мыши обнаруживают отсоединенную TM с отсутствием tectorins, подобно у мышей Tecta
ΔENT/ΔENT. Гетерозиготные Tecta
Y1870C/+ мыши обнаруживают разрушенную и частично истонченную TM, которая экспрессирует tectorins и всё-ещё частично соединена с Кортиевым органом (Figure 2, A-C). Хотя взаимодействия между гетерозиготными TM и OHC ,по-видимому, нормальные с почти нормальным транспортом обратной связи от OHC к BM, но чувствительность к звуковым сигналам снижена из-за подъёма естественных порогов активации. Пространство между TM и IHC увеличено у гетерозигот, а движения IHC и reticular lamina специфически снижены при характерных частотах. Т.о., гетерозиготные Tecta Y1870C/+ мыши помогают предположить вторую роль для TM: хотя пучки IHC волосков не внедрены непосредственно в TM, TM всё ещё выполняет роль по передаче вибраций BM к IHC при определенных частотах. Др. словами, TM подгоняет вибрации BM, чтобы оптимально стимулировать IHC при своих наилучших частотах(Legan et al., 2005). Эта гипотеза была недавно подтверждена физиологическими исследованиями, подтвердившими, что пучки волосков из IHC движутся в ответ на движения жидкости в узком пространстве между IHC и TM. Эти движения жидкости возникают в результате вибрации TM и движений пучков волос OHC (Nowotny and Gummer, 2006). В то время как мутации в TECTA уже были связаны с NSHL у людей (Hughes et al., 1998; Verhoeven et al., 1998; Mustapha et al., 1999), но мутации TECTB ещё не были обнаружены у лиц с нарушениями слуха. Однако нокаутные мыши по beta-tectorin недавно были описаны. Хотя матрикс TM у гомозиготных мышей нарушен, их внутреннее ухо менее чувствительно только к тонам низких частот, тогда как к высоким частотам чувствительность отсутствует или снижена (sharpness cochlear tuning). Эти результаты подтверждают третью роль TM: влиять на cochlear frequency resolution (Russell et al., 2007).
Intra-hair bundle link proteins: Cdh23, Pcdh15, Vlgr1 and Ush2a
Синдром Usher имеет более общую этиологию для комбинации наследственно глухоты и слепоты. Это заболевание комбинирует врожденную нейросенсорную потерю слуха и прогрессирующую потерю поля зрения из-за retinitis pigmentosa (RP), который ведет к прогрессирующей дегенерации сетчатки. Описаны три клинических субтипа синдрома Usher. Эти типы отличаются временем начала и характером потери слуха, началом RP и вовлечением вестибулярной дисфункции [rev. (Nikolopoulos et al., 2006)]. До сих пор мутации 9 генов были сцеплены с синдромом Usher у людей 5 из этих генов были также сцеплены с NSHL у людей: MYOVIIA, USH1C/Harmonin, CDH23, PCDH15 и VLGR1/MASS1 (Van Camp and Smith, 2006). Сегодня получены мутантные мыши для 8 из сцепленных с Usher генов. Белки. кодируемые Usher генами, принадлежат к разным классам и имеют разные функции. Однако все эти белки участвуют в молекулярной функции, развитии и/или поддержании пучков волосковых клеток. Недавно было установлено, что все связанные с Usher белки связаны ( непосредственно или косвенно) др. с др. посредством harmonin's PDZ сайтов и формируют мультипротеиновую единицу, которая может перемещаться взад-вперед (посредством моторных миозинов myosin VIIa и/или myosin XVa) вдоль актиновых филамент волосковых клеток к месту своего действия внутри стереоцилий (Figure 1F) [rev. (Kremer et al., 2006; Reiners et al., 2006)]. 4 связанных с Usher гена кодируют адгезивные белки (cadherin 23, CDH23; protocadherin 15, PCDH15; Very Large G-protein coupled Receptor-1, VLGR1/ MASS1; и usherin, USH2A). Др. связанные с Usher гены кодируют внутриклеточные белки волосковых клеток (Supplementary Table S1). Описана и роль связанных с Usher белков в глазах (Reiners et al., 2006). Стереоцилии являются высоко специализированными микроворсинками с актиновым стержнем, которые проецируется от волосковых клеток в эндолимфатическое пространство. В пучке волосков стереоцилии собраны в упорядоченные ряды со специальным лестнице-образным паттерном ( Figure 1, D-F). У мышей развитие пучков волосков происходит в эмбриогенезе и в течение первых двух недель после рождения. С Usher-связанные адгезивные белки участвуют в связках между стереоцилиями, существенными для механотрансдукции, процесса, с помощью которого кохлеарные и вестибулярные волосковые клетки транслируют механические движения своих пучков волосков в электрохимические сигналы. Во внутреннем ухе млекопитающих развитие и созревание пучков волосков существенно отличается (for details, Figure 1E legend). Cadherin 23 (известный также как otocadherin, Cdh23) и protocadherin 15 ( Pcdh15) являются трансмембранными белками с коротким внутриклеточным и длинным внеклеточным доменом, они являются атипичными членами cadherin сверхсемейства. Все молекулы cadherin содержат cadherin домены ('EC' домены) вдоль своей внеклеточной части, которая обеспечивает Ca2+-зависимую димеризацию cadherin молекул. Димеризация cadherin белков от двух соседних клеток связывает клетки [rev. (Reiners et al., 2006)]. На своем цитоплазматическом конце, cadherin 23 и protocadherin 15 содержат класса I-PDZ binding (PBM) мотивы, которые могут связывать PDZ- содержащие белки. Следовательно, они могут связывать harmonin. Посредством harmonin связанные с Usher cadherins оказываются сцепленными с цитоскелетными актиновыми филаментами и оказываются частью с Usher связанной мультибелковой единицы (Siemens et al., 2002; Adato et al., 2005b)]. Во внутреннем ухе мышей дикого типа cadherin 23 локализуется на стереоцилиях волосковых клеток и на Reissner's мембране (Wilson et al., 2001; Boeda et al., 2002; Lagziel et al., 2005). В пучках волосков мышей cadherin 23 располагается вдоль длины растущих стереоцилий и на кончиках зрелых стереоцилий. Точнее, cadherin 23 локализуется на связках между стереоцилиями в пучке волосков (Boeda et al., 2002; Siemens et al., 2002; Siemens et al., 2004; Lagziel et al., 2005; Michel et al., 2005; Rzadzinska et al., 2005). Дв сплайс-варианта Cdh23 обнаружены во внутреннем ухе мышей. Оба имеют PDZ-связывающие мотивы, которые могут связывать harmonin. Описан укороченный cadherin 23, лишенный внеклеточного домена [rev. (Reiners et al., 2006)]. Protocadherin 15 широко экспрессируется во многих тканях мышей (Alagramam et al., 2001a; Murcia and Woychik, 2001) и людей(Alagramam et al. , 2001b), включая головной мозг, улитку и преддверие, начиная с раннего развития и в течение всего периода жизни. В развивающейся улитке protocadherin 15 локализуется на апикальной поверхности волосковых клеток, поддерживающих клеток, клеток наружной бороздки и клеток спирального ганглия, тогда как взрослая улитка экспрессирует protocadherin 15 только в стереоцилиях волосковых клеток (Alagramam et al., 2001b). Ботльшинство мутантных по Cdh23 мышей жизнеспособно. 4 различные мутации в Cdh23 возникли спонтанно у мышей: waltzer (Deol, 1956; Di Palma et al., 2001a; Wilson et al., 2001; Lagziel et al., 2005), waltzer niigata (Wada et al., 2001), modifier of deafwaddler - mdfw (Bryda et al., 2001) и age-related hearing loss - Ahl (Noben-Trauth et al., 2003). Инъекции химических соединений самцам мышей были использованы для получения Cdh23 мутантного потомства. И chlorambucil, индуцирующий делеционные мутации [Albany-waltzer (Bryda et al., 1997)], и ENU, индуцирующий точечные мутации (три типа waltzer-Jackson аллелей; описаны в Mouse Genome Database: http://www.informatics.jax.org) дополнили Cdh23 мутантных мышей. 7 из этих Cdh23-мутантных линий мышей (исключая Ahl) обладают сходным фенотипом: NSHL с поведением кружения, качанием головы и беспорядочными движениями, которые появляются у гомозигот с рождения. Гетерозиготы выглядят нормально при рождении, но обнаруживают тенденцию к прогрессирующей потере слуха в более зрелом возрасте и обнаруживают высокую чувствительность к индуцируемой шумами потере слуха (Holme and Steel, 2004). Аллель Ahl является естественно возникшим Cdh23G753A диморфизмом, появившимся во многих лаб. инбредных линиях мышей. Замещение guanosine 753 на adenosine выщывает in-frame пропуск экзона 7, это приводит к тенденции развития прогрессирующей потери слуха во время старения и к высокой чувствительности к вызванной шумами потере слуха (Davis et al. , 2001; Noben-Trauth et al., 2003). Waltzer мутантные мыши обладают прогрессирующей дезорганизацией пучков волосков, которая впервые обнаруживается в начале образования пучков на ст. эмбриогенеза 18.5 (E18.5) и становится выраженной, когда волосковые клетки созревают (Figure 2, Q-R). Кроме того, kinocilium размещается неправильно. В более старом возрасте стереоцилии утолщаются и сливаются, приводя к дегенерации волосковых клеток (Di Palma et al., 2001a; Wada et al., 2001; Holme and Steel, 2002). C57BL/6J мыши, которые гомозиготны по Ahl аллелю обнаруживают дегенерацию волосковых клеток в апикальной части улитки. OHC затрагиваются больше, чем IHC. Наблюдается также дегенерация эфферентных нервных волокон (Mizuta et al., 1993). В развивающихся волосковых клетках внутреннего уха мышей cadherin 23 располагается как в kinocilial, так и временных боковых связках (Boeda et al., 2002; Lagziel et al., 2005; Michel et al., 2005), но waltzer мутантный cadherin 23 отсутствует только в боковых связках. Cadherin 23 обнаруживается также вдоль киноцилий зрелых вестибулярных волосковых клеток (Lagziel et al., 2005). Волосковые клетки Cdh23-дефицитных рыбок данио лишены кончиковых связок и у этих рыб наблюдаются дефекты баланса и слуха (Sollner et al., 2004). Установлено, что cadherin 23 у мышей является также компонентом кончиковых связок между стереоцилиями кохлеарных и вестибулярных пучков волосков. Более того, cadherin 23 обладает биохимическими свойствами, сходными с теми, что кончиковых связках. Следовательно, cadherin 23, по-видимому, составляет кончиковые связки, которые регулируют механически открываемые ионные каналы в стереоцилиях волосковых клеток (Goodyear and Richardson, 2003; Siemens et al., 2004). Первой мышиной моделью мутантного Pcdh15 была Ames-waltzer ( av). Ames-waltzer мыши были описаны ещё в 1956, как несущие спонтанную рецессивную мутацию, вызывающую глухоту. поведение кружения, трясение головой и гиперактивность, сходная с waltzer ( v) фенотипом (Schaible, 1956). В последующие годы было найдено независимо в том же самом локусе несколько мутаций, дающих сходный фенотип. Мутантный ген был найден в Pcdh15 в Ames-waltzer аллеле, который возник у трансгенных мышей вследствие инсерционного мутагенеза (Alagramam et al., 1999). Поведение кружения и снижение потребления краски AM1-43, как было установлено, коррелировало с нормальной функцией трансдукции в волосковых клетках, и со структурными дефектами в преддверии, которые обнаруживались с помощью световой или сканирующей электронной микроскопии. Функциональный дефект приводил к дезорганизации стереоцилий в улитке и saccule, это приводило к дисфункции волосковых клеток и к прогрессирующей дегенерации. Т.к. внутреннее ухо на ст. P10 у гомозигот обнаруживало только аномальные стереоцилии в улитке, saccular стереоцилии начинали обнаруживать признаки дезорганизации только на P30, а во внутреннем ухе взрослых гомозиготных мышей (P50 или старше) наблюдалась практически полная дегенерация кортиева органа улитки и вестибулярной saccular macula (как поддерживающие, так и волосковые клетки отсутствовали). В улитке наблюдается вторичная дегенерация нейронов спирального ганглия. Нейроэпителий в utricle и cristae полукружных каналах выглядит нормальным, но utricular otoconia были крупными аномальной формы (Alagramam et al., 1999; Alagramam et al., 2001a; Alagramam et al., 2005). У др. спонтанных Pcdh15 мутантов, возникших в результате инсерции остатка цитозина, которая привела к сдвигу рамки и образованию преждевременного стоп-кодона, фенотип был очень схожим, хотя мыши были не полностью глухими. Дезорганизация кохлеарных стереоцилий наблюдается у новорожденных (P0) (Hampton et al. , 2003). ENU-индуцированные Pcdh15 мутантные мыши имеют сходный фенотип, дезорганизацией стереоцилий, но не прежде ст. P2. В улитке IHC оказывались менее затронут ыми по сравнению с OHC (Washington et al., 2005). Три модели, описанные выше являлись гомозиготами по функциональным нулевым аллелям. Более умеренные фенотипы описаны у мышей, гомозиготных по менее тяжелым мутациям в Pcdh15 (Pawlowski et al., 2006). В волосковых клетках внутреннего уха мышей экспрессируется некоторые изоформы protocadherin 15 и два из них, как полагают, являются частью комплексов связок кончиков и киноцилий в пучках волосков Др. изоформа может быть ассоциирована с временными боковыми связками между развивающимися стереоцилиями и связками к киноцилиям, т.к. паттерн экспрессии этих изоформ сходен с таковым cadherin 23 (Ahmed et al., 2006).
Ген Vlgr1/Mass1 у мышей транскрибируется в несколько сплайс-вариантов, которые кодируют интегральные и секретируемые белки. Самая длинная из изоформ, Vlgr1b, которая приблизительно размером в 19 kb, транслируется в самый крупный из известных белков клеточной поверхности (~ 6300 fvbyjrbckjn), содержащий крупный внеклеточный домен. Его внутриклеточный домен содержит PBM мотив, который может взаимодействовать с harmonin's PDZ доменом. Хотя белок Vlgr1b обладает типичной структурой G-protein coupled рецептора с 7 трансмембранными доменами, его функция неизвестна (McMillan et al., 2002; Yagi et al., 2005). Mass1 является самым маленьким (~ 9400 оснований) сплайс-вариантом Vlgr1. Vlgr1 рецепторы экспрессируются преимущественно в нейроэпителии развивающегося головного мозга (Yagi et al., 2005), а Vlgr1 мутации, в особенности мутантные Vlgr1b и Mass1 транскрипты оказываются ассоциированными с аудиогенными судорагами у мышей (Skradski et al., 2001; McMillan and White, 2004; Yagi et al., 2005) и у людей (Nakayama et al., 2002). Внеклеточные домены Vlgr1 рецепторов содержат множественные повторяющиеся единицы CalX-β модули, которые связывают Ca2+ катионы и могут играть роль в Ca2+-зависимой межклеточной адгезии. Предполагается. что эти модули ммогут отслеживать уровни внеклеточного Ca2+ и участвовать в трафике внутри- и внеклеточного Ca2+ (Nikkila et al., 2000; Weston et al., 2004). Дополнительные мотивы во внеклеточных доменах белков Vlgr1, как полагают. взаимодействуют с др. связанными с Usher белками [rev. (Reiners et al., 2006)]. Первая мышиная модель мутантов Vlgr1 была Mass1Frings, которая возникла спонтанно в 1951 (Frings et al., 1951), она служит в качестве мышиной модели эпилепсии, обусловленной чувствительностью к судорогам на громкие шумы. BUB/BnJ инбредная линия мышей является гомозиготной по Mass1Frings мутации и обладает как аудиогенными судорогами, так и прогрессирующей потерей слуха, которая начинается постнатально и прогрессирует до полной глухоты (Zheng et al., 1999; Skradski et al. , 2001). BUB/BnJ мыши являются также гомозиготами по Ahl аллелю Cdh23, но этот факт не объясняет глухоты у всех этих мышей, т.к. в др. линиях, гомозиготных по Ahl вероятность и тяжесть потери слуха значительно ниже. Ассоциация VLGR1 мутаций с HHL включает синдром Usher type II у людей (Weston et al., 2004) , это открывает возможность того, что FringsMass1 мутация лежит в основе потери слуха у BUB/BnJ мышей, В самом деле, было показано, что совместные мутации Cdh23 и Vlgr1 ответственны за более тяжелую потерю слуха у BUB/BnJ мышей. У молодых BUB/BnJ мышей кохлеарные стереоцилии развиваются аномально и остаются незрелыми. Стереоцилии разъединены и отсоединены, временами обнаруживаются вне их единицы, а в наиболее тяжело поврежденных пучках теряют свою полярность и градированную высоту. На более старых возрастах волосковые клетки и клетки спиральных ганглиев дегенерируют (Johnson et al., 2005). Экспрессия Vlgr1 рецепторов дикого типа во внутреннем ухе ограничивается регионом синапсов и стереоцилиями волосковых клеток, как в преддверии, так и улитке. В волосковых клетках Vlgr1 рецепторы экспрессируются только в основании развивающихся стереоцилий в той же самой локализации и в то же время, что и ankle связки: их экспрессия максимальна в перинатальный период и снижается во время развития волосковых клеток. Моноклональные антитела, которые используются для идентификации ankle связок у эмбрионов кур, как было установлено, связывают птичий ортолог Vlgr1b. Получены две мышиные модели с мутантным Vlgr1: (a) нокаутные мыши, которые не экспрессируют Vlgr1 белков (Yagi et al., 2005) и (b) Vlgr1/del7TM мыши, у которых целенаправленная делеция была использована для удаления трансмембранного домена Vlgr1 (McGee et al., 2006).
В обеих моделях недостаток Vlgr1 рецепторов приводит к сходным аномалиям улитки. Гомозиготные мыши погибают, не обладая ankle связками между стереоцилиями волосковых клеток. Хотя пучки волосков выглядят нормальными при рождении, они становятся дезорганизованными позднее. Мыши, гомозиготные по мутантному Vlgr1 обнаруживают выраженную глухоту на третьей неделе жизни и с этого возраста обнаруживают дезорганизованные пучки волосков, включая смещение киноцилия, это ведет к искаженному развитию стереоцилий. Т.о., Vlgr1 рецепторы, как полагают, являются критическим членом комплекса ankle связок. Неожиданно, хотя развивающиеся вестибулярные пучки волосков содержат ankle связки и экспрессируют Vlgr1, но только кохлеарные волосковые клетки повреждены у гомозиготных мышей. Вестибулярные клетки не дегенерировали и отсутствовали вестибулярные симптомы у пациентов с Usher II (McGee et al., 2006; Yagi et al., 2007). Др. интегральный Usher-related белок, usherin (кодируемый длинным транскриптом
Ush2a), также, как полагают, является компонентом ankle связок в развивающихся стереоцилиях (Adato et al., 2005a). У людей мутации
USH2A ответственны за наиболее распространенную генетическую форму синдрома Ushe (Eudy et al., 1998). Сходным образом нокаутные Ush2a
-/- мыши обладают прогрессирующей дегенерацией фоторецепторных клеток, их слух повреждается лишь слегка, присутствует умеренная не прогрессирующая HI на высокие частоты. Хотя usherin, как полагают, является частью ankle белка и обнаруживается в основном в основании развивающихся стереоцилий во внутренних и наружных волосковых клетках (со ст. E20) вдоль всей улитки,
Ush2a-/- мыши имеют нормальные пучки волосков и теряют только OHC в базальном витке улитки (Adato et al., 2005a; Liu et al., 2007). Это указывает на то, что экспрессия молекул связок в раннем развитии пучков волосков является существенной для их корректного образования и созревания. Правильное созревание пучков волосков является критическим для жизнеспособности волосяных клеток.
Genes responsible for endolymph production
Улитка содержит два раздельных заполненных жидкость компартмента с разными концентрациями ионов (Figure 1A). Перилимфатическое пространство содержит перилимфу, раствор с высоким Na+ и низким K+, сходный с др. внеклеточными жидкосятями тела. Верхушки волосковых клеток обращены в эндолимфу, которая имеет противоположный состав катионов, высокий K+ и низкий Na+, тогда как их базолатеральная поверхность купается в перилимфе. Циркуляция катионов калия в улитке из перилимфы в эндолимфу через кохлеарную боковую стенку и поддержание уникального ионного состава эндолимфы являются существенными для функции слуха. Изучение мышиных моделей по мутантным белкам, которые участвуют в рециклинге K+ в улитке помогло установить механизм рециклинга. Акустически вызываемые рецепторные потенциалы генерируются притоком ионов K из эндолимы в волосковые клетки. Эти K+ ионы затем секретируются базолатерально во внеклеточное пространство Кортиева органа и поднимаются с помощью поддерживающих клеток. После этого ионы K+ транспортируются латерально в направлении спиральной лигаменты посредством щелевых соединений между поддерживающими клетками и от поддерживающих клеток к корневым (root) клеткам, высвобождаясь во внеклеточное пространство спиральной лигаменты и затем с помощью второй сети щелевых соединений между соединительнотканными фиброцитами катионы переносятся в направлении stria vascularis. Ионы K+ проходят через базальную мембрану между соединительной тканью и эпителиальными клетками stria vascularis посредством плотных соединений и высвобождаются из эпителиальных базальных клеток во внеклеточное пространство stria vascularis. Затем маргинальные клетки stria vascularis принимают ионы K+ и высвобождают их обратно в эндолимфу. Маргинальные клетки stria vascularis и Deiters' клетки, как пример поддерживающих клеток проиллюстрированы на Figures 1C и 1D, соотв. Сходный путь рециклинга существует в преддверии. Это описание несколько упрощено, т.к. некоторое протекание K+ из эндолимфы осуществляется через наружные sulcus клетки и Reissner's мембрану [rev. and illustrated in (Kikuchi et al., 2000; Wangemann, 2002)]. Некоторые гены, которые ответственны за HHL у людей кодируют белки, которые участвуют в циркуляции K+ в улитке. Мутантные модельные мыши были получены для следующих генов: (a) Gjb2/ Cx26, Gjb6/Cx30 и Cldn14, которые кодируют белки межклеточной адгезии: гены Gjb2 иd Gjb6 кодируют белки щелевых соединений connexin 26 (Cx26) и connexin 30 (Cx30), тогда как Cldn14 кодирует белок плотных соединений; (b) Kcne1, Kcnq1 и Kcnq4, которые кодируют каналы ионов калия; и (c) Slc26a4, которые кодируют транспортеры анионов. Щелевые соединения являются каналами, соединяющими две клетки и обеспечивающие быстрый транспорт широкого разнообразия ионов и малых молекул (включая нуклеотиды, siRNAs и inositol phosphates) между соединенными клетками. Щелевые соединения состоят из плотно соединенных внутримембранных канальных частиц (коннексонов), которые в свою очередь являются гексамерными ансамблями коннексиновых белков. Волосковые клетки внутреннего уха не содержат щелевых соединений. Две самостоятельные сети щелевых соединений существуют в улитке: между клетками соединительной ткани и между несенсорными эпителиальными клетками. Cx26 и Cx30 являются частью как кохлеарной системы щелевых соединений, так и образуют ансамбли, чтобы сформировать гибридные (гетеромерные) щелевые соединения. Однако превалирующей изоформой коннексина, экспрессирующейся в поддерживающих клетках улитки является Cx26 (Ahmad et al., 2003; Forge et al., 2003; Buniello et al., 2004). В геноме человека, GJB2 и GJB6 гены располагаются в одном и том же хромосомном локусе ( DFNB1, 13q11-12). Мутации в этом локусе отвечают за большую пропорцию врожденной наследственной NSHL с изменчивостью зависящей от популяции [~ 30-60%; e.g. (Zelante et al., 1997)]. GJB2 мутации являются наиболее частым источником глухоты у людей (30-50% случаев доречевой наследственной NSHL). В большинстве таких случаев ответственными мутациями являются маленькие делеции в гене GJB2 , а тип наследования аутосомно рецессивный. Немногие случаи доминантно наследуемой SHL обусловлены мутациями GJB2. До сих пор более сотни связанных с глухотой различных мутаций в GJB2 было идентифицировано у людей. Крупные делеции в GJB6 гене также могут вызывать глухоту у гомозигот. Кроме того, комбинация крупной делеции в GJB6 и точковая мутация в GJB2 могут вызывать NSHL у гетерозигот [Connexins and Deafness Homepage; http://davinci.crg.es/deafness/ (Ballana et al., 2007)].
Два разных подхода, целенаправленный мутагенез (Gabriel et al. , 1998) и ENU-индуцированный мутагенез (Coghill et al., 2002), используются для нокаута гена Gjb2/Cx26 у мышей. Оба подхода ведут к рождению только хорошо слышащих гетерозиготных потомков, гомозиготные эмбрионы погибают in utero из-за дефектов плаценты. Две дополнительные стратегии предприняты для получения мутантных Gjb2 мышиных моделей, которые были бы жизнеспособны и имели нарушения слуха. Gjb2 был специфически нокаутирован в эпителиальной сети улитки (поддерживающие и фланкирующие эпителиальные клетки), используя условную cre-loxP систему, чтобы генерировать мышей, которые гомозиготны по Gjb2-loxP и несут Cre после промотора Otog, который экспрессируется только в эпителиальных клетках улитки (Figure 2, D-F) (Cohen- Salmon et al., 2002). Во втором подходе целенаправленный точечный мутагенез использован для репликации Cx26 R75W мутации (Kudo et al., 2003), которая ответственна за аутосомно доминантную SHL (HHL и болезнь кожи) у гетерозиготных людей (Richard et al. , 1998). Доминантное наследование объясняется способностью мутантного Cx26 ингибировать функцию щелевых соединений, которые собираются совместно из дикого типа и мутантных Cx26 молекул (Richard et al., 1998). Как Gjb2 нокаутные гомозиготы, так и Cx26 R75W
гетерозиготы обладают сходной HI у взрослых и гистологическим фенотипом, хотя вторая модель имеет более тяжелый фенотип. В обеих моделях развитие внутреннего уха нормальное вплоть до постнатального дня 14 (P14). Только после начала восприятия звуков на ст. P15-P16 эпителиальные клетки начинают гибнуть от апоптоза. Соседствующие с IHC поддерживающие клетки повреждались первыми. После этого OHC и поддерживающие их клетки начинали гибнуть. Кортиев канал спадается. Cx26 R75W гетерозиготы обнаруживают дегенерацию всего органа Корти, которая начинается на ст. P14 и ведет к полной дегенерации как волосковых клеток, так и поддерживающих клеток в возрасте 7 недель. У Gjb2 нокаутных мышей, IHC погибают только при наиболее выраженном нарушении слуха у мышей (но если они выживают, то обнаруживаются незрелые синапсы) , а некоторые intradental клетки спирального limbus дегенерировали в более старом возрасте (P60). Ретикулярная ламина на апикальной поверхности сенсорного эпителия, которая состоит из плотных соединений между волосковыми клетками и поддерживающими их клетками, разрушается с ранних стадий у Gjb2 нокаутных мышей (Figure 2, E- F). Следовательно, Cx26, по-видимому, существенен для выживания и функционирования Кортиева органа, но не обязателен для его нормального развития. Различия между моделями наблюдались в поддержании различий электрического потенциала между компартментами эндолимфы и перилимфы в улитке, отражающимися в виде endocochlear potential (EP). У Gjb2 нокаутных мышей эндолимфатическая концентрация K+ и EP были значительно ниже у гомозиготных мышей, как и ожидалось, это подтверждает гипотезу, что базирующиеся на Cx26 щелевые соединения необходимы для рециклинга K+ в улитке. Неожиданно, EPs у Cx26 R75W гетерозигот были нормальными, указывая тем самым, что причиной апоптоза клеток в Кортиевом органе в присутствие мутации Cx26 является нарушение транспорта K+ поддерживающими клетками скорее, чем нарушением гомеостаза эндолимфы, как это и предполагалось. Т.к. Cx26 не был нокаутирован в преддверии в conditional модели и его вестибулярная экспрессия была нормальна у гомозиготных мышей, то эти мыши не обладали вестибулярными дефектами. Однако не было обнаружено ни вестибулярных, ни др. аномалий и во второй модели. Кроме того, хотя доминантная мутация Cx26 R75W экспрессируется также в клеточной системе соединительной ткани улитки, не обнаруживаются видимые структурные изменения в stria vascularis или спиральной лигаменте (Cohen-Salmon et al., 2002; Kudo et al., 2003).
Gjb6 нокаутные модельные мыши были получены с помощью инсерции missense мутации. Гомозиготные мыши были жизнеспособны и плодовиты, но имели нарушения слуха и были лишены EP. Дегенерация Кортиева органа, обусловленная апоптозом, наблюдалась сл стадии P18, также как и у Gjb2 мутантных мышей (Teubner et al., 2003).
Cx26 и Cx30 образуют ко-ансамбли в одних и тех же щелевых соединениях (Ahmad et al., 2003; Forge et al., 2003). Хотя Cx30 не способен формировать гомомерные щелевые соединения в Cx26-дефицитных клетках, и Cx30 не может компенсировать отсутствие Cx26 в conditional нокаутной модели (Cohen-Salmon et al., 2002). Разные коннексины отличаются размерами и ионной избирательностью и обладают характерными voltage-gating чувствительностями. Как результат коннексоны собираются из разных коннексинов, они обладают разной проницаемостью и пропускными функциями (Bruzzone and Cohen-Salmon, 2005; Zhao et al., 2006)]. Т.о., характеристики гомомерных коннексонов, собранных только из Cx30 могут быть отличными от гетеромерных коннексонов, собираемых из Cx26 и Cx30. Даже если проницаемость для малых ионов (подобных K+) сходна в коннексонах разного типа, доставка более крупных вторичных messenger молекул может быть отличной, и может влиять на приток K+ косвенно. Предполагается. что некоторые Gjb2 мутации влияют на проницаемость щелевых соединений для inositol triphosphate скорее, чем K+. Неспособность рециклинга K+ из поддерживающих клеток обратно в эндолимфу, как полагают, является вторичной по отношению к транспорту inositol triphosphate (Beltramello et al., 2005). Несмотря на это неспособность Cx30 компенсировать отсутствие Cx26 может возникаить в результате низкой экспрессии. В противоположном случае избыточная экспрессия Cx26 у Gjb6 нокаутных мышей полностью восстанавливает слух и предупреждает дегенерацию волосковых клеток. Т.о., по крайней мере, Cx26 может компенсировать отсутствие Cx30, подтверждая, что гетеромерные щелевые соединения, которые содержат Cx26 и Cx30, не существенны для нормального слуха и для жизнеспособности Кортиева органа у мышей. Интересно, что Gjb6 нокаутные мыши недоэкспрессируют Cx26 белок в улитке, подтверждая ускоренную деградацию гомомерных щелевых соединений. Gjb6 нокаутные мыши, которые также несут ген для повышенной экспрессии Cx26, экспрессируют избыточно Cx26 в печени, но в улитке уровни Cx26 нормальны, подтверждая, что гомомерные Cx26 щелевые соединения менее стабильны. чем гетеромерные Cx26-Cx30 ансамбли, но обладают сходной функцией (Ahmad et al., 2007).
Хотя connexin 29 (Cx29) не участвует в рециклинге K+ ионов в улитке, его следует упомянуть, т.к. мутации в гене GJE1/Cx29 обнаружены у NSHL пациентов (Yang et al. , 2007). Распределение Cx29 в улитке сильно отличается от Cx26 и Cx30. В отличие от Cx26 и Cx30, которые в основном экспрессируются в поддерживающих клетках и фиброцитах улитки, Cx29 экспрессируется в основном в Швановских клетках спирального ганглия и на низких уровнях в stria vascularis (Eiberger et al., 2006; Tang et al., 2006b). Экспрессия Cx29 в головном мозге и др. органах также в основном приходится на миэлинирующие клетки. Получены нокаутные Gje1 мыши. Одна группа не обнаружила аномалий у Cx29-дефицитных C57BL/6 мышей, включая нормальные слои миэлина (Eiberger et al., 2006), др. группа описала потерю слуха из-за сильной демиэлинизации в строме нейронов спирального ганглия (neuropathy), с пенеттрантностью ~50% и отсутствием повреждений в нейроэпителии внутреннего уха у BALB/c мышей (Tang et al., 2006b).
Плотные соединения, наиболее апикальные соединения эпителиальных клеток, служат в качестве основного ион-селективного барьера против околоклеточного переноса жидкости. Кроме того, они вносят вклад в поддержание клеточной полярности, образуя внутримембранный барьер, который ограничивает латеральную диффузию апикального и базолатерального компонентов мембран. Плотные соединения состоят, по крайней мере, из трех типов трансмембранных белков: occludin, claudins и членов семейства junction adhesion molecule (JAM). Более 20 claudins известно, каждый из которых имеет характерную проницаемость [rev. (Kondoh et al., 2006)]. В улитке основное разделение перилимфы от эндолимфы достигается с помощью плотных соединений, которые запечатывают пространства между клетками, ограничивающими жидкие компартменты. После идентификации рецессивных мутаций CLDN14 у человека как ответственных за выраженную NSHL у людей (Wilcox et al., 2001), были получены Cldn14-нулевые мыши, чтобы выяснить роль claudin 14 во внутреннем ухе. Claudin 14 был обнаружен в плотных соединениях ретикулярной ламины улитки (плотные соединения между волосковыми клетками м поддерживающими клетками и между соседними поддерживающими клетками). Cldn14-нулевые мыши имели нормальный EP, но были глухи. Вестибулярных отклонений не обнаруживалось. Хотя плотные соединения ретикулярной ламины микроскопически выглядят нормально у Cldn14-нулевых мышей, стереоцилии волосковых клеток потеряны или дезорганизованы во время первых трех недель жизни, за тем быстро наступает дегенерация волосковых клеток. OHC дегенерируют раньше IHC. Т.к. claudin 14 обладает более высокой проницаемостью для K+, чем для Na+ он может быть необходим для поддержания собственно ионного состава перилимфатической жидкости, окружающей базолатеральную поверхность OHC. Правильный ионный состав жидкости может быть существенным для жизнеспособности OHC (Ben-Yosef et al., 2003).
Гены Kcne1, Kcnq1 и Kcnq4 кодируют субъединицы низким напряжением активируемых калиевых каналов, которые являются основными детерминантами клеточной реполяризации в возбужденных клетках. Они открываются в ответ на деполяризацию и облегчают избирательный отток K+ через плазматическую мембрану. Каждый канал состоит из 4-х alpha и нескольких beta субъединиц. Т.к. формирующие пору alpha субъединицы достаточны, чобы сформировать функциональный канал, beta субъединицы детерминируют уникальные свойства каналов, включая одноканальное проведение, общую активность канала, voltage зависимость, временную зависимость активации, чувствительность к температуре и pH, а также чувствительность к лекарствам [rev. (Wangemann, 2002)]. Маргинальные клетки stria vascularis и вестибулярные темные клетки секретируют K+ в эндолимфу только с помощью K каналов, состоящих из Kcnq1 (alpha) и Kcne1 (beta) субъединиц. Следовательно, Kcnq1/Kcne1 каналы ответственны за образование эндолимфы (Marcus et al., 1997; Neyroud et al., 1997; Marcus et al., 1998; Nicolas et al., 2001). В кардиальных миоцитах Kcnq1/Kcne1 K+
каналы несут медленно активируемый выпрямитель (rectifier) K+ тока, который играет основную роль в фазе реполяризации кардиального потенциала действия. Следовательно, мутации в KCNE1 или KCNQ1 у людей индуцируют неотличимые SHL фенотипы (Jervell and Lange-Nielsen Syndrome) HHL и кардиальных симптомов, включая удлиненные QT интервалы и аритмии , сопровождаемые syncope или внезапной смертью (Neyroud et al., 1997; Schulze-Bahr et al., 1997; Tyson et al., 1997).
Kcne1 (Vetter et al., 1996; Nicolas et al., 2001) или Kcnq1 (Lee
et al., 2000; Casimiro et al., 2001; Rivas and Francis, 2005) нокаутные мыши характеризуются классическим waltzer-подобным фенотипом с тяжелой потерей слуха и вестибулярными симптомами, вплоть до полной глухоты у взрослых мышей. Хотя гистология внутреннего уха нормальна при рождении, изменения развиваются позднее. Strial маргинальные клетки и вестибулярные темные клетки были неспособны секретировать K+ ионы, это вело ко вторичной дегенерации нейроэпителия, включая волосковые клетки и коллапс эндолимфатического пространства. Сходным образом эндолимфатическое пространство спадается у пациентов с Jervell and Lange-Nielsenсиндромом (Friedmann et al., 1966). При рождении EP по сравнению с мышами дикого типа очень низок с высокими концентрациями Na+ и низкими K+ в эндолимфе. После рождения EP постепенно увеличивается (особенно после P7), достигает взрослых значений на ст. P14 (Yamasaki et al., 2000). Соответственно, Kcne1 (Vetter et al., 1996) или Kcnq1 (Casimiro et al., 2001) нокаутные мыши обнаруживают нормальное эндолимфатическое пространство при рождении. Лишь спустя 3 дня после рождения спадается Reissner's мембрана и уменьшается эндолимфатическое пространство. Спонтанная точковая мутация в Kcne1 также возникла у (punk rocker мышей; Kcne1 pkr). Гомозиготные мыши, экспрессирующие сильно укороченный Kcne1 белок, имеют сходный фенотип с тем. что у Kcne1 нокаутных мышей (Letts et al., 2000). Kcnq1 нокаутные мыши обнаруживают также дефекты кардиальной реполяризации (Casimiro et al., 2001; Casimiro et al., 2004). Т.к. Kcnq1 является сердцевиной канала, то очевидно, что Kcne1 необходим для доставки его в плазматическую мембрану, т.к. вестибулярные темные клетки у Kcne1 нокаутных мышей экспрессируют Kcnq1 в своей цитоплазме скорее, чем в их апикальных мембранах (Nicolas et al., 2001). Т.о., Kcne1, по-видимому, важен для доставки в мембрану Kcnq1 и/или стабильности Kcnq1 в мембране.
Kcnq4 является alpha субъединицей M-типа K+ канала. M-типа каналы являются очень медленными voltage-зависимыми K+ каналами. В нейронах М-каналы могут противостоять устойчивой деполяризации мембран и повторным затуханиям потенциалов действия после сильного возбуждающего импульса, но они также могут временно повышать возбудимость нейронов после воздействия на них модуляторных нейротрансмиттеров (Cooper and Jan, 2003). Соотв., Kcnq4 каналы были обнаружены в нейронах некоторых ядер центрального аудиторного пути. Однако Kcnq4 обнаружен также в базолатеральной мембране волосковых клеток улитки (Beisel et al., 2000) и преддверия (Rocha-Sanchez et al., 2007) мышей ( OHC и IHC). После начала восприятия слуха (P12-14), он локализуется исключительно на базальном полюсе. Поэтому было предположено, что Kcnq4 каналы ответственны за секрецию излишка K+ ионов из волосковых клеток в перилимфу, окружающую их базолатеральную мембрану, и за регулировку потенциала покоящихся мембран волосковых клеток (Kharkovets et al., 2000; Boettger et al., 2002; Beisel et al., 2005; Rocha-Sanchez et al., 2007). У людей, KCNQ4 мутации вызывают аутосомно доминантную NSHL, подтверждая тем самым, что мутантный ген оказывает доминантный негативный эффект, когда он ко-экспрессирует с аллелем дикого типа (Kubisch et al. , 1999). Две мышиные модели с мутантным Kcnq4 получены: гомозиготные нокаутные мыши и knock-in мыши с точковой мутацией, которая имитирует доминантно негативную мутацию людей. Вестибулярные симптомы отсуствовали в обепх моделях, хотя Kcnq4 экспрессируется на высоком уровне в вестибулярных волосковых клетках WT. Мыши имеют нормальный слух на постнатальных стадиях, но начинается прогрессирующая потеря слуха, которая сопровождается прогрессирующей дегенерацией OHC. Прогрессирование глухоты и потери OHC было более быстрым у гомозиготных knockout и knock-in мышей (недели) по сравнению с гетерозиготными knock-in мышами (месяцы). Использование селективного ингибитора Kcnq каналов для выделения Kcnq-зависимых K+ токов, не было выявлено Kcnq-зависимых K+ токов в OHC у гомозигот или доминантно негативных гетерозиготных мышей, это приводило к деполяризованным потенциалам покоящихся мембран в OHC. IHC практически не затрагивались. Поэтому было предположено, что мутации Kcnq4 индуцируют прогрессирующую HHL из-за хронической деполяризации OHC, приводя к их дегенерации (Kharkovets et al., 2006). Было установлено, что экспрессия Kcnq4 в регулируется с помощью тироидного гормона. Thyroid hormone receptor TRa непосредственно влияют на экспрессию Kcnq4 во время финальной дифференцировки OHC. У TRa 1 нокаутных мышей Kcnq4 экспрессируется, но аномально распределяется как в базальных, так и латеральных частях мембран OHC (Winter et al., 2006).
SLC26 (solute carrier protein 26) семейство анионных обменников включает интегральные белки с 10-12 трансмембранными доменами, которые могут транспортировать некоторые анионы, включая chloride, iodide, sulfate, nitrate, bicarbonate, hydroxyl, oxalate и formate. Каждый член этого семейства обладает храктерным сродством и специфичностью на каждый анион. Два члена SLC26 оказаличь связанными с HHL у людей: SLC26A4/pendrin и SLC26A5/prestin. SLC26A4 мутации были ассоциированы как с SHL (Pendred syndrome) (Everett et al., 1997) , так и с NSHL (Li et al., 1998; Usami et al., 1999), тогда как SLC26A5 были ассоциированы только с NSHL (Liu et al., 2003). Pendred Syndrome, впервые был описан в 1896 (Pendred, 1896), он характеризуется нейросенсорной глухотой и увеличенным тироидным зобом с повышенной iodine разрядкой после воздействия perchlorate. Большинство пациентов обладало также радиологически определяемыми структурными уродствами внутреннего уха, наиболее распространенным признаком которых было увеличение вестибулярного эндолимфатического протока [rev. (Glaser, 2003)]. Увеличение эндолимфатического протока наблюдалось также у пациентов с NSHL, обусловленной мутациями в SLC26A4 (Li et al., 1998; Usami et al., 1999). В гетерологичной системе экспрессии pendrin, как было установлено, транспортирует iodide, chloride, formate и nitrate (Scott et al., 1999; Scott and Karniski, 2000). Используя мышей и крыс, было установлено, что pendrin экспрессируется в апикальных мембранах клеток щитовидной железы, почек и внутреннего уха. Отсутствие pendrin, как полагают, непосредственно ответственно за дефектную организацию iodide у Pendred пациентов. Однако нокаутные Slc26a4 мыши лишены тироидных симптомов (Everett et al., 2001) и точная роль pendrin в щитовидной железе до сих пор неясна. Во внутреннем ухе мышей pendrin выявляется в апикальных мембранах клеток, покрывающих эндолимфатическую полость, которые рассматриваются как играющие важну роль в гомеостазе эндолимфы (Everett et al. , 1999; Royaux et al., 2003; Yoshino et al., 2004). Кроме того экспрессия pendrin в улитке обнаруживается также в поддерживающих клетках Кортиева органа (Claudius и Deiters' клетки), а также в спиральной лигаменте и спиральном ганглии. Недпавно с помощью postembedding immunogold анализа в ЭМ была выявлена некоторая экспрессия pendrin также в OHC и IHC, в частности в их апикальных мембранах и стереоцилиях (Yoshino et al., 2006).
Slc26a4 нокаутные мыши (Pds) обладают waltzer-подобной вестибулярной дисфункцией и полной глухотой. Внутреннее ухо у них развивается нормально вплоть до ст. E15, два дня спустя после начала экспрессии pendrin у мышей дикого типа. Затем формируются тяжелые расширения эндолимфатической полости как в улитке, так и преддверии (Figure 2, K-L). Эт а дилятация, как полагают, вторична по отношению к измененным осмотическим условиям и повышению объема эндолимфатической жидкости. Во время второй постнатальной недели волосковые клетки начинают дегенерировать. В преддверии otoconia и otoconial мембраны также деструктуированы (Everett et al., 2001). После прекращения вскармливания маргинальные клетки stria vascularis Pds-/- мышей обнаруживали нерегулярные формы и размеры, что приводило к истончению stria vascularis. У взрослых Pds-/- мышей гиперпигментация клеток stria vascularis предшествовала их дегенерации, это указывает на повреждения свободными радикалами. Функциональные эксперименты показали, что Pds-/- мыши постепенно теряют EP, начиная с P12, перед тем как начинается обычно восприятие звуков. Несмотря на это концентрация эндолимфатического K
+ и экспрессия Kcnq1/Kcne1 каналов были нормальными. Pendrin недостаточность устранялась экспрессией Kcnj10 K
+ каналов в strial промежуточных клетках, хотя Kcnj10 мРНК экспрессировалась нормально (Royaux et al., 2003; Wangemann et al., 2004). Kcnj10 каналы играют роль в рециклинге K
+ ионов через базальноклеточный барьер в stria vascularis. Т.о., pendrin может выполнять роль по поддержанию EP, не затрагивая секреции K
+ из маргинальных клеток stria vascularis, а скорее за счет нарушения истечения K
+ в промежуточных клетках.
Kcnj10 нокаутные мыши не генерируют EP, но имеют уменьшенные эндолимфатический объем и концентрацию K
+ (Marcus et al., 2002). Следовательно, дефицит pendrin может иметь дополнительные результаты. Др. роль pendrin была недавно выявлена как в улитке (Wangemann et al., 2007) , так и преддверии (Nakaya et al., 2007). Ca
2+ каналы (Trpv5 и Trpv6) в эпителиальных клетках преддверия и улитки резорбируют ионы кальция из эндолимфы и ингибируются низким pH. В улитке Trpv5 и Trpv6 экспрессируется в маргинальных клетках stria vascularis и sulcus эпителиальных клетках, соотв. Эти каналы поддерживают низкие концентрации Ca
2+ нормальной эндолимфы. Pendrin-нокаутные мыши обладают низким pH и более высокой концентрацией Ca
2+ в эндолимфе, приводя к снижению трансэпителиального потенциала в utricle. Высокий уровень Ca
2+ в эндолимфе может ингибировать сенсорную трансдукцию, необходимую для восприятия звуков и способствовать дегенерации волосковых клеток. Т.о., во внутреннем ухе pendrin действует как Cl
-/HCO
3, который обеспечивает секрецию щелочных HCO
3 ионов в эндолимфатическое пространство и одной из его возможных ролей может быть поддержание в эндолимфе pH (Nakaya et al., 2007; Wangemann et al., 2007). Гиперпигментация stria vascularis у взрослых Pds
-/- мышей позволяет предположить, что воспалительный процесс участвует в их дегенерации. В самом деле, эта гиперпигментация и реорганизация маргинальных клеток происходят конкурентно с инвазией макрофагов особенно в stria vascularis, и с экспрессией макрофаговых и complement маркеров (Jabba et al., 2006). Ген winged helix/forkhead
Foxi1 (известный также как Fkh10 ) , как полагают, индуцирует экспрессию pendrin, т.к.
Foxi1-нулевые мыши не экспрессируют pendrin и обладают сходным фенотипом с таковым у нокаутных по pendrin мышей (Hulander et al., 2003).
SLC26A5/prestin - the motor protein of outer hair cell electromotility
Улитка млекопитающих характеризуется двумя механизмами амплификации звуковых сигналов: (a) амплификация движений стереоцилий с помощью mechano-electric трансдуцирующих каналов (существует во всех известных аудиторных органах); и (b) OHC somatic electromotility - voltage-зависимые быстрые изменения в длине и жесткости OHC (существует только во внутреннем ухе млекопитающих), наз. также как кохлеарный усилитель (amplifier). Electromotility включает укорочение деполяризованных OHC и удлинение гиперполяризованных клеток, независимо от уровня АТФ или OHC Ca
2+ . Амплификация с помощью OHC electromotility ? как полагают, амплифицирует кохлеарные вибрации и делает возможной повышенную остроту слуха и избирательность частот улитки млекопитающих. Этот механизм делает возможной реакцию улитки на низкие (менее 1 KHz) частотные сигналы [rev. (Frolenkov, 2006)]. Prestin является интегральным белком, который экспрессируется только в OHC улитки ( OHC см Figure 1D). Prestin молекулы, как мономеры. так и тетрамеры обильно экспрессируются вдоль боковых мембран OHC и в меньшей степениt - в базальной части мембраны. Онтогенетическая экспрессия prestin совпадает с появлением electromotility (Belyantseva et al., 2000; Zheng et al., 2000; Yu et al., 2006). Хотя prestin принадлежит к семейству SLC26 обменников анионов и имеет сходную структуру с др. членами этого семейства, четкие доказательства, показывают, что он действует как транспортер ионов? еще не представлены. Более того, нокаут prestin ( Slc26a5) у мышей не затрагивает характерные для всей клетки токи OHC (Liberman et al., 2002). Вместо этого prestin рассматривается как voltage-зависимый моторный белок, ответственный за OHC electromotility (Zheng et al., 2000), т.к.
Slc26a5 нокаутные мыши не обнаруживают OHC electromotility (Figure 2M) и избирательности частот. Эти мыши подтверждают гипотезу, что OHC electromotility усиливает чувствительность внутреннего уха, т.к. они характеризуются 40-60 dB потерей кохлеарной чувствительности при отсутствии нарушений в пучках волосков OHC и механо-электрической трансдукции. Кроме того,
Slc26a5-нулевые мыши обладают более короткими (Figure 2N), это не удивительно, т.к. prestin очень обилен в боковых стенках этих клеток. CD возрасте 4-9 недель наблюдается вторичный апоптоз OHC в базальной четверти улитки у
Slc26a5-нулевых мышей, сопровождаемый дегенерацией IHC, хотя IHC не экспрессируют prestin. Однако HI предшествует дегенерации волосковых клеток, по крайней мере, за две недели, это значит, что отсутствие electromotility является первичной причиной потери слуха (Liberman
et al., 2002; Cheatham et al., 2004; Wu et al., 2004). Недавно было установлено, что типичное распределение prestin вдоль боковых мембран OHC зависит от thyroid hormone receptor TRβ
(Winter et al., 2006). хотя абсолютная величина OHC electromotility у гетерозиготных мышей вдвое ниже нормальной (Liberman et al., 2002), функция и вид улитки у мышей только с одной копией гена
Slc26a5 нормальны (Cheatham et al., 2005). Было предположено, что prestin ощущает эл. напряжение путем связывания внутриклеточных ионов C
l- в деполяризованных клетках. В результате его конформация меняется. Т.о., prestin является очень эффективным прямым voltage-to-force конвертором. Его функция связана с типичной нелинейной ёмкостью (capacitance), которая может быть измерена [ rev. (Dallos et al., 2006)].
Unconventional myosins
Неконвенционные миозины являются моторными молекулами, которые содержат actin-связывающий домен на своем N-терминальном моторном или головном домене. Используя АТФ в качестве источника энергии они могут перемещаться вдоль актиновых филамент. Нестандартные миозины имеют также сайты связывания для белков на своих С-терминальных хвостах и поэтому они могут служить в качестве "машин", которые перевозят белки грузы к их местам мишеням в клетке. Во внутреннем ухе млекопитающих экспрессируется несколько нестандартных миозинов, каждый из которых имеет уникальный паттерн экспрессии и функцию во внутреннем ухе. Мутации в 5 генах миозинов (
Myo1a, Myo3a, Myo6, Myo7a и Myo15a) оказались связанными с HHL у людей. паттерн экспрессии миозина 1A во внутреннем ухе мышей ещё не изучен. Myo3a, Myo6, Myo7a и Myo15a экспрессируются ов внутреннем ухе мышей только в волосковых клетках и играют роль в организации пучков волосков [rev. (Hertzano and Avraham, 2005)]. Два из них, миозины VIIa и XV, могут связывать PDZ сайты на harmonin или whirlin и являются частью Usher-related сети, которая проиллюстрирована на Figure 1F [rev. (Reiners et al., 2006)]. Так, myosin VIIa (Boeda et al., 2002; Senften et al., 2006) и myosin XVa (Belyantseva et al., 2005) активно транспортируют harmonin и whirlin, вместе с прикрепленными белками к соотв. местам на стереоцилиях. Недавно было установлено, что myosin IIIa также локализуется на кончиках стереоцилий и необходим для их собственного поддержания (Schneider et al., 2006). Мышиные модели доступны только для мутаций в Myo6, Myo7a и Myo15a. Нулевые мутации в Myo6 [Snell's waltzer (Avraham et al., 1995)], Myo7a [shaker1 (Self et al., 1998)] и в Myo15a [shaker2 (Probst et al., 1998)] вызывают сходные waltzer-подобные фенотипы у гомозигот (глухота и вестибулярные дисфункции) из-за слияний ( Myo6), дезорганизации ( Myo7a) или укорочения (Myo15a) стереоцилий.
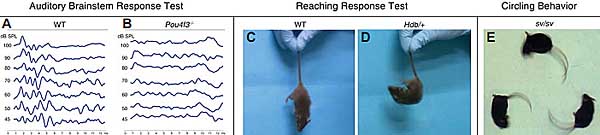
Myo6 и Myo7a нулевые мыши также обнаруживают последующую дегенерацию волосковых клеток. В то время как мыши гомозиготные по нулевым мутациям в Myo6, Myo7a или Myo15a были глухи, гетерозиготы имели нормальный фенотип. Более того, двойные гетерозиготные мыши по Myo15a и др. ( Myo6 или Myo7a) нулевому аллелю также были нормальными (Karolyi et al., 2003). Однако missense Myo7a мутация (headbanger мыши; Hdb) вызывали вестибулярный фенотип и легкую HI также у гетерозигот (Figure 3D), из-за элонгации и слияния стереоцилий волосковых клеток. Гомозиготы обладали более тяжелым фенотипом (Rhodes et al., 2004). Myo6 может быть рассмотрен более детально в качестве примера. Спонтанная мутация, Snell's waltzer (sv), возникла в 1966 (Deol and Green, 1966). Поведение кружения Snell's waltzer мышей представлено на Figure 3E. Индуцированная радиацией мутация в том же самом локусе также доступна (se
sv/se
sv) (Russell, 1971), как и индуцированный ENU мутант (ENU89) (personal communication, Colin Fletcher and Karen Avraham). Мутация в гене
Myo6 найдена в
sv аллеле (Avraham et al., 1995). Во внутреннем ухе мышей и рыбок данио myosin VI экспрессируется специфически в апикальной плазматической мембране волосковых клеток, вблизи основания стереоцилий (Self et al., 1999; Kappler et al., 2004). Гомозготные
sv мыши обладают прогрессирующей дегенерацией волосковых клеток внутреннего уха со ст. P12, это ведет к дегенерации всего нейроэпителия внутреннего уха. Ранние стадии развития волосковых клеток и стереоцилий были нормальными, т.к. при рождении только часть пучков волосков была дезорганизована. Однако во время первой недели постнатального развития пучки волосков оказывались дезорганизованными и апикальная плазматическая мембрана волосковых клеток оказалась приподнятой. Затем во время следующих двух недель стереоцилии были аномально слиты вместе, чтобы сформировать гигантские не функциональные стереоцилии (Self et al., 1999; Kappler et al., 2004). Сходный фенотип наблюдался у рыбок данио с мутацией в гене
Myo6b. У рыбок данио ген
Myo6 удвоен ( Myo6a и Myo6b) и только
Myo6b преимущественно экспрессируется во внутреннем ухе и нейроэпителии боковой линии. Подобно
sv мышам мутации
Myo6b у рыбок данио ответственны за слуховые и вестибулярные дефекты (
satellite мутанты) из-за дезорганизации пучков волосков, в которых стереоцилии в конечном итоге сливаются. Структурные дефекты в апикальной плазматической мембране тоже обнаруживаются, как и крупные пузырьки, которые накапливаются вблизи кутикулярной пластинки (Seiler et al., 2004). Базируясь на мутациях
satellite у рыб и
sv у мышей, было предположено, что myosin VI прикрепляет апикальную плазматическую мембрану стереоцилия к стержневым актиновым филаментам (Figure 1F). В отсутствие myosin VI, апикальная плазматическая мембрана возвышается над эпителием и между стереоцилиями, приводя к слиянию стереоцилий. Мутации в гене
MYO6 человека оказались связанными с HHL только спустя 6 лет после идентификации мутаций
Myo6 у
sv мышей. В то время как у мышей Myo6 мутации были ассоциированы только с рецессивной NSHL, мутации
MYO6 у людей были связаны как с доминантными (Melchionda et al., 2001) , так и рецессивными NSHL (Ahmed et al., 2003), а также с доминантными SHL, которые включали кардиальную гипертрофию и удлинение QT в дополнение к нейросенсорной HHL (Mohiddin et al., 2004).
Hair cell genes for transcription factors
Сенсорные волосковые клетки и не сенсорные поддерживающие клетки в нейроэпителии внутреннего уха возникают из общего предшественника. Просенсорные клетки предшественники дифференцируются в волосковые клетки по умолчанию, но это решение к дифференцировке в целом ингибируется передачей сигналов Notch (Yamamoto et al., 2006). Активация Notch латерально репрессирует экспрессию транскрипционного фактора Math1/Atoh1, который совместно с Sox2 необходим, чтобы индуцировать дифференцировку просенсорных клеток предшественников в волосковые клетки. В самом деле, Atoh1 нокаутные мыши не имеют волосковых клеток в преддверии и улитке (Bermingham et al., 1999; Yamamoto et al., 2006). Множество дополнитеьных транскрипционных факторов являются критическим для развития внутреннего уха. Некоторые из них специфически экспрессируются в волосковых клетках внутреннего уха (напр. Pou4f3) , а др. являются критическими и для развити др. органов тоже. Некоторые транскрипционные факторы коррелируют с глухотой у людей и мышей( Eya1, Pou3f4, Pou4f3, Mitf, Pax3, Snai2/Slug, Sox2, Sox10, Six1 ; see Supplementary Table S1). Sox2 и Pou4f3 будут описаны в
SOX2 мутации у людей коррелируют в основном с билатеральной anophthalmia (уродствами глаз) у гетерозигот (Fantes et al. , 2003). Однако, две новых мутации de novo SOX2 коррелируют с SHL у гетерозигот. Мутация nonsense (Q155X), как полагают, ответственна за HI, в дополнение к anophthalmia, отсутствие всех оптических путей и др. нейрологические аномалии (Hagstrom et al., 2005); а missense мутация (479delA), как полагают. ответственна за синдром комбинированного врожденного hypothalamo-pituitary нарушения и HI (Kelberman et al., 2006). У эмбрионов мышей Sox2 экспрессируется в основном в развивающейся ЦНС и сенсорных плакодах, где он играет критические роли в эмбриогенезе. На ст. E9.5, Sox2 экспрессируется не только в нервной трубке, но и также в отоцисте, из которого развивается нейроэпителий внутреннего уха. В развивающейся улитке Sox2 обычно экспрессируется только в просенсорных клетках предшественниках, а также в дифференцированных волосковых клетках и поддерживающих клетках развивающегося органа Корти (Wood and Episkopou, 1999; Kiernan et al., 2005). Два мутантных Sox2 аллеля, Lcc (light coat and circling) и Ysb (yellow submarine), были получены у мышей с помощью соотв. мутагенеза, используя рентгеновское облучение или инсерцию трансгена, соотв. Мыши, несущие мутантные аллели, легко идентифицируются, из-за полудоминантной желтой окраски шерсти. Аллели Lcc и Ysb содержат интактными кодирующие и соседние последовательности Sox2, но регуляторные элементы, которые затрагивают экспрессию Sox2 мутантны (вставка последовательности использована для получения аллеля Ysb, содержащего регуляторную последовательность от гена Col2a1). В результате Lcc и Ysb гомозиготные E9.5 эмбрионы мыши экспрессируют нормальный Sox2 в нервной трубке, но не экспрессируют (Lcc) или less (Ysb) Sox2 в отоцисте. Т.о., мутации не нарушают развития головного мозга, включая умеренный фенотип по сравнению с мутациями SOX2, которые были описаны у людей. Гомозиготные мыши обладают тяжелой HI ( Ysb мыши) или полной глухотой(Lcc мыши), а также поведением кружения из-за уродства внутреннего уха и его нейроэпителия. А преддверие затрагивается более тяжело. При рождении, Lcc мыши, которые не экспрессируют Sox2 в своем внутреннем ухе, обладают более тяжело нарушенными внутренними ушами, а нейроэпителий отсутствует полностью. т.к. волосковые и поддерживающие клетки неспособны дифференцироваться. Ysb которые экспрессируют низкие уровни Sox2 во внутреннем ухе, практически не имеют волосковых клеток в преддверии. В базальной области улитки, Ysb гомозиготы обнаруживают аномальные участки дезорганизованных волосковых клеток с регионами, не содержащими волосковых клеток между ними (Figure 2, G-J). Апикальная область их улитки включает дезорганизованные волосковые клетки с отсутствием четкого расчленения на IHC и OHC. Уникальный паттерн развития волосковых клеток у Ysb мышей может быть результатом инсерции Col2a1 регуляторного мотива в регуляторную последовательность Sox2. Lcc гомозиготы, которые не обнаруживают экспрессии Sox2 во внутреннем ухе, не экспрессируют Atoh1, тогда как Ysb гомозиготы, которые экспрессируют всё-таки низкие уровни Sox2 экспрессируют и Atoh1. Следовательно, Sox2, как и предполагалось, действует выше Atoh1 (Dong et al., 2002; Kiernan et
Гены POU-доменовых транскрипционных факторов, как известно, контролируют терминальные стадии развития ЦНС [rev. (Ryan and Rosenfeld, 1997)]. У мышей, Pou4f3 (известный также как Brn-3c или Brn3.1) экспрессируется совершенно специфически в волосковых клетках улитки(Figure 2O) и преддверия. Его экспрессия может быть обнаружена в волосковых клетках внутреннего уха со ст. E12.5, после начала экспрессии
Atoh1
и постепенно увеличивается вплоть до рождения (Xiang et al., 1998; Hertzano et al., 2004). Мутация
POU4F3 сцеплена с аутосомно доминантной прогрессирующей NSHL у людей (Vahava et al. , 1998).
Pou4f3-нокаутные мыши (Erkman et al., 1996; Xiang et al. , 1997), также как и dreidel (
ddl) мыши, которые не экспрессируют функционального Pou4f3 (Hertzano et al., 2004), обладают сходными waltzer-подобными фенотипами выраженной глухоты (Figure 3, A-B) и вестибулярной дисфункцией, включая трясение головы, поведение кружения и гиперактивность.
Pou4f3-нокаутные мыши характеризуются прогрессирующей потерей волосковых клеток как в преддверии, так и улитке, это ведет ко вторичной дегенерации поддерживающих клеток, а также к дегенерации нейронов спирального и вестибулярного ганглиев.
Pou4f3 экспрессируется в постмитотических клетках просенсорных предшественников, которые предетерминированы к развитию волосковых клеток, но не в предетерминированные митотические клетки. Волосковые клетки в развивающемся внутреннем ухе
Pou4f3 нокаутных мышей испытывают первоначальную дифференцировку, но не способны формировать зрелые стереоцилии, некоторые из волосковых клеток неправильно локализованы в слое поддерживающих клеток и все они или большинство из них деградируют с помощью апоптоза во время стадии поздней беременности или ранних постнатальных дней. Т.о., Pou4f3 является критическим для нормальной терминальной дифференцировки, миграции и выживания волосковых клеток внутреннего уха (Erkman et al., 1996; Xiang et al. , 1997; Xiang et al., 1998). Гены транскрипционных факторов Gfi1 и Lhx3 как полагают, являются мишенями для Pou4f3. Гомозиготные
dreidel мыши экспрессируют минимальные уровни мРНК Gfi1 в волосковых клетках улитки и преддверия (Figure 2P) и не экспрессируют
Lhx3 в волосковых клетках улитки (Figure 2, S-V) (Hertzano et al., 2007).
Deaf mouse mutants not correlated with human hereditary hearing loss
Мутантные модельные мыши не получены для всех генов, которые были связаны с HHL у людей. Из 61 клонированного гена, которые были ассоциированы с HHL людей (Van Camp and Smith, 2006), 18 не имеют мышиных моделей до сих пор (Table 2). Как упоминалось во введении, 75% генов, которые были сцеплены с нарушениями или дисфункциями внутреннего уха у мышей (Jackson_Laboratory, 2007) пока не удается связать с HHL у людей (Van Camp and Smith, 2006). Продукты некоторых из этих генов могут взаимодействовать с уже известными связанными с глухотой сетями. Напр., лизосомный мембранный белок Scarb2/LIMP-2, который регулирует транспорт в мембрану некоторых белков, был опознан как существенный для локализации Kcnq1/Kcne1 калиевых каналов в апикальной мембране маргинальных клеток stria vascularis и вестибулярных темных клеток у взрослых мышей. Как результат, Scarb2-дефицитные мыши обнаруживают прогрессирующую потерю слуха из-за дегенерации stria vascularis (Knipper et al., 2006). Др. гены, которые были сцеплены с HI у нокаутных мышей, представляют класс продуктов, которые ещё не связаны с HHL у людей и являются критическими для функции внутреннего уха. Напр., creatine kinase (Ckb) может играть роль в переносе АТФ на ATPases стереоцилий.
Ckb нокаутные мыши обладают HI и вестибулярной дисфункцией. Цитозольная brain изоформа creatine kinase является наиболее многочисленным белком после β-actin в пучках волосков utricle птиц, как показывает mass-спектрометрия (Shin et al., 2007). Др. примером является спонтанная мутация (jbg), которая ведет к HI и вестибулярной дисфункции у
jitterbug мышей и картируется в гене
Clic5. Clic5 принадлежит ксемейству хлорных межклеточных каналов. Во внутреннем ухе мышей он специфически обнаруживается в базальных частях стереоцилий волосковых клеток улитки и преддверия. Jitterbug мыши обладают аберрантными стереоцилиями и прогрессирующей дегенерацией волосковых клеток, указывающие на то, что Clic5 может играть роль в сборке и поддержании стереоцилий. Clic5, по-видимому, ассоциирует с radixin в основаниях стереоцилий и , по-видимому, участвует в формировании или стабилизации соединений между плазматическими мембранами стереоцилий и их актиновой сердцевиной (Gagnon et al., 2006). Одна и та же мутация может индуцировать разные фенотипы у разных инбредных линий мышей, которые экспрессируют разные генетические модификаторы. Такие модификаторы описаны также у людей [первым стал локус
DFNM1 (Riazuddin et al., 2000)]. У мышей этот феномен известен как эффект влияния фона. Напр.,
mdfw и Ahl аллели гена
Cdh23 могут индуцировать возраст-зависимую и индуцируемую шумами потерю слуха у гомозигот некоторых линий мышей (с разным временем начала в разных линиях), но др. линии относительно устойчивы к этим мутациям (Zheng and Johnson, 2001; Noben-Trauth et al., 2003). Мутантные аллели Cdh23 могут действовать как генетические модификаторы у гетерозигот. Т.о., аллель
Ahl может модифицировать потерю слуха у Mass1
frings мутантных мышей (Johnson et al., 2005). По крайней мере, 7 дополнительных локусов ммогут индуцировать зависимую от возраста потерю слуха у мышей. У двугенных мутантов мышей, которые несут два мутантных гена и обнаруживают разные фенотипы по сравнению с гомозиготами по мутации в одном из генов, один из мутантных генов может рассмтариваться как ген модификатор [e.g. (Adato et al., 1999; Johnson et al., 2005; Zheng et al., 2005)]. Чтобы идентифицировать такие модифкаторы, некоторые группы скрещивали мышей, несущих связанные с глухотой мутации, с мышами из разных линий [e.g. (Asher et al., 1996; Niu et al., 2006)]. См. обзор (Johnson et al., 2006).
Summary
Efforts are now underway to create knock-outs and conditional mutants for every gene in the mouse genome [NIH knockout mouse project, (KOMP): http://www.nih.gov/science/models/ mouse/knockout/; and European conditional mouse mutagenesis program (EUCOMM): http://www.eucomm.org/]. This endeavor will undoubtedly create many more mouse models for human HHL. As discussed above, there have been many cases where the mouse gene has led to the discovery of the human HI gene (and vice versa), emphasizing the complementarity of mouse and human studies in the auditory and vestibular systems. Complex hearing impairment, which includes both noise-induced hearing loss and age-related hearing loss (presbyacusis), as well as the identification of modifiers, will require additional mouse models.
The identity of a human mutation is critical for human diagnostics and genetic counseling, and early identification and intervention is beneficial for hearing impaired patients (White, 2004; Hyde, 2005). The information acquired from mouse morphological and physiological studies, as exemplified from the various techniques in Figure 2, demonstrates that the study of mouse models for deafness will undoubtedly provide a key to understand auditory function and help develop critical elements for therapeutics (Atar and Avraham, 2005; Tang et al., 2006a).
Сайт создан в системе
uCoz