Гипофизарный ACTH (adrenocorticotropic hormone, corticotropin) является первичным регулятором синтеза надпочечного глюкокортикоида (Gc). Хотя он чувствителен к большому набору стимулов, велючая стрессы, уровни ACTH в плазме тонко контролируются для поддержания равновесия hypothalamo-pituitary-adrenal (HPA) оси. Последствия дисбаланса НРА осуществляются посредством Gc и оказывают распространенное метаболическое влияние. Гипофизарный ACTH, следовательно, является центральным регулятором этой оси. В работе обсуждаются регуляторные механизмы продукции ACTH, с упором на дефекты, которые вызывает дефицит или избыток гормона. ACTH продуцируется с помощью протеолитического расщепления предшественника про-гормона, pro-opiomelanocortin (POMC). Ген РОМС т. о., ответственен за синтез ACTH и является единственной копией гена, которая кодирует различные биологически активные пептиды, включая α-melanotropin (αMSH) и β-endorphin. Хотя гипофиз является первичным местом экспрессии РОМС, этот ген экспрессируется также в ряде гипоталямических нейронов, которые являются критическими для гомеостаза энергии. Интересно отметить, что межвидовые различия в продукции αMSH из РОМС. В самом деле, в то время как гипофиз грызунов имеет промежуточную долю, состоящую из меланотрофных клеток, в которых РОМС превращается в αMSH, люди теряют промежуточную долю гипофиза во время беременности и основной источник αMSH для пигментации продуцируется локально в коже. Хотя и важные, но эти отличия не представляют собой очень высокую консервацию регуляторных механизмов, контролирующих продукцию АСТН.
Ранние события органогенеза гипофиза рассматриваются на этих стр. (1) вместе с лежащими в основе молекулярными механизмами. Транскрипционные регуляторы, ответственные за дифференцировку клонов соматотрофов (продуцирующих гормон роста, GH), тиротрофов (продуцирующих thyrotropin) и лактотрофов ( продуцирующих prolactin), которые как известно включают Pit1 (2,3) и Prop1 (Prophet-of-Pit)(4). Однако, взаимоотношения между этими клонами и теми, что ответственны за продукцию АСТН и гонадотропины, были в основном неизвестны. Орфановый ядерный рецептор SF1 участвует в поздней дифференцировке гонадотрофов (продуцирующих gonadotropins luteinizing hormone и follicle-stimulating hormone). Мало известно о дифференцировке кортикотрофинов (продуцирующих АСТН proteolitic cleavage of POMC) и меланотрофов (продуцирующих αMSH вследствие более глубокого процессинга РОМС). Открытие Tpit, критического регулятора дифференцировки РОМС клона пролило новый свет на взаимоотношения между клоном РОМС и гонадотрофами и привело также к реализации плохо определяемого клинически заболевания, isolated ACTH deficiency (IAD). Параллельно получена информация о механизме регуляции Gc негативной обратной связи гипофизарного РОМС, подтвердившая механизм Gc резистентности в кортикотрофных аденомах, которые вызывают болезнь Cushing.
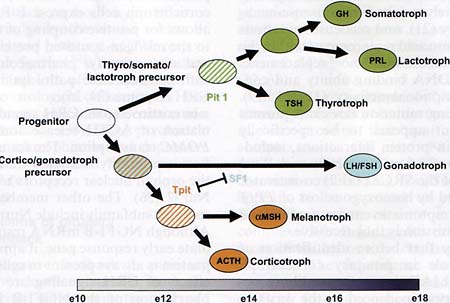
Fig/ 1. Differentiation of pituitary cells. This binary model of cell differentiation is largely derived from genetic studies of the
critical regulators of lineage specificity such as Pitl, Tpit and SFl. The undifferentiated progenitors as well as the sublineage
precursors have been proposed as a result of these studies but never isolated. The growth hormone (GH), prolactin (PRL) and
thyroid-stimulating hormone (TSH) producing cells are presumed to have a Pitl-dependent common thyro-somato-lactotroph precursor. This precursor is distinct from a putative cortico-gonadotroph precursor that was proposed as result of cell fate changes between gonadotropin- (LH/FSH) and POMC-expressing lineages. Two POMC lineages express the pro-
hormone gene that is either processed into corticotropin (ACTH) in anterior lobe corticotrophs or into α-melanotropin
(αMSH) in intermediate lobe melanotrophs. The differentiation scheme is aligned with a mouse developmental time line indicating the time of appearance of each cell type. FSH, follicle-stimulating hormone; LH, luteinizing hormone; POMC, pro-opiomelanocortin.
Pituitary cell differentiation and the corticotroph fate
Все клетки переднего и промежуточного гипофиза происходят из инвагинации ротового эпителия, известного как карман Ратке. Дорсальная часть кармана Ратке развивается в интимном контакте с проекцией из диэнцефалона, infundibulum, и эта область кармана дает промежуточную долю, которая имеет очень гомогенный клеточный состав и целиком состоит из меланотрофов у грызунов. Помимо этой дорсо-вентральной асимметрии, различные исследования на мышах выявили градиенты сигнальных молекул, экспрессируемых вокруг и внутри развивающегося гипофиза в течение всего раннего развития (5). Также экспрессия многих критических транскрипционных факторов, экспрессируемых во время раннегог развития, является асимметричной внутри развивающейся железы, что указывает на молекулярную гетерогенность клеток несмотря на их внешнее сходство. Следовательно, возможно, что клетки становятся детерминированными в специфичные клоны или субнаборы клонов раньше, чем выявляются доступные сегодня маркеры. Сегодня самые ранние маркеры для каждого из клонов являются ограниченные клетками транскрипционные факторы, которые были первоначально идентифицированы по их роли в клеточно-специфичной транскрипции соотв. гормональных генов. Т.о, транскрипционный фактор Pit1 маркирует клетки клонов соматотрофов, тиротрофов и лактотрофов (5). В соответствии с ограниченной экспрессией Pit1 в этих клонах недостаточность PIT1 ведет к дефициту этих трех клонов (6,7). Те же самые клоны маркируются также несколько раньше экспрессией Prop1 и сходным образом мутации в PROP1 ведут к дефициту тех же самых клонов у людей (7) и мышей (8). Мутации обоих этих факторов проявляются карликовостью, т.к. в каждом случае имеется дефицит продукции GH. Клон гонадотрофов маркируется экспрессией SF1? а мутации SF1 ведут к дефициту гонадотропина (9); однако, этот дефицит может быть скорректирован инъекциями gonadotropin-releasing hormone, указывая тем самым, что клетки гонадотрофы присутствуют и что дефицит является ограничивающим экспрессию гена гонадотропина (10). Исследования Pit1 и Prop1 ясно показали наличие общего предшественника для трех клонов, которые зависят от этих факторов, взаимоотношения между гонадотрофами и Pit1- зависимыми клонами или с РОМС- экспрессирующими клонами оставались неясными до тех пор, пока Т-бокс фактор Tpit не был идентифицирован в РОМС клоне.
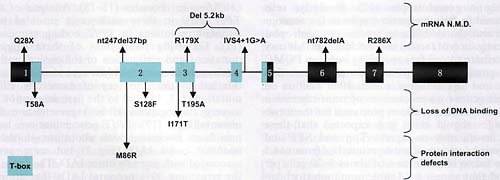
Fig. 2. Human TPIT mutations responsible for neonatal onset isolated ACTH deficiency (IAD). Until discovery of TPITas a causative gene for IAD, this condition was poorly recognized and thought to be extremely rare. Neonatal onset IAD is now recognized as a very homogenous clinical entity, and two-thirds of these cases are caused by loss-of-function TPIT gene mutations. This recessive condition is associated with nonsense mutations that lead to nonsense mRNA decay (NMD). A number of missense mutations within the T-box domain (DNA binding domain) of TPIT cause loss of transcriptional activity as a result of the loss of the DNA binding ability. Finally, one very interesting mutation, M86R also within the T-box, is deficient in protein protein interactions; this mutation prevents homodimerization of Tpit, its interaction with Pitxl and with the co-activator TIF2 (SRC2).
Специфичная для гипофиза транскрипция гена РОМС критически зависит от Tpit, который сильно ограничен в экспрессии кортикотрофными и меланотрофными клетками (11). В соотв. с этой ограниченной экспрессией мутации потери функции Tpit у мышей вызывают тяжелый дефицит в экспрессирующих РОМС и кортикотрофных клетках (12). Нехватка Tpit не полностью предупреждает дифференцировку кортикотрофов и меланотрофов, т.к. дефицитные мыши всё ещё имеют небольшие количества РОМС-экспрессирующих клеток и это приводит к чрезвычайно низкому уровню в плазме АСТН и Gc, ассоциирующему с гипоплазией надпочечника и недоразвитием кортекса (13). Вторичная недостаточность надпочечников, возникающая в результате дефицита АСТН, наблюдаемого у таких мышей, очень сходна по клиническим проявлениям IAD человека.
Помимо демонстрации позитивной роли Tpit в дифференцировке кортикотрофов и меланотрофов,
Tpit нокаутные мыши также четко выявляют тесное взаимотноешие между этими клонами и гонадотрофами. В самом деле, промежуточная доля у
Tpit-/- мышей содержит значительные количества клеток, которые имеют измененную клеточную судьбу, чтобы стать аутентичными гонадотрофами (12). Сходным образом, клетки, предназначенные стать кортикотрофами передней доли, также изменяют судьбу, чтобы стать гонадотрофами. Исследования факторов и механизмов, которые могут вносить вклад в преключение клеточной судьбы между РОМС и гонадотрофами, привело к демонстрации, что Tpit и SF1 противодействуют др. др. в отношении генов мишеней посредством механизма транс-репрессии (12). Если и в самом деле общий предшественник для этих двух клонов существует, то следует ожидать, что эти предшественники могут ко-экспрессировать Tpit и SF1 и сканирование срезов целых гипофизов от Е14.5 мышей привело к идентификации приблизительно 5-20 клеток на железу, которые, по-видимому, обладают двойной иммунохимией, являясь позитивными по обоим факторам. в согласии с гипотезой общего предшественника. Эта работа привела к предложению бинарной модели для дифференцировки клеток гипофиза (Рис. 1).
Tpit in isolated ACTH deficiency
Критическая роль Tpit для экспрессии РОМС указывает на то, что их мутации у людей д. вызывать IAD. IAD редкое заболевание, которое часто ассоциирует в воспалительным заболеванием гипофиза, такими как hypophysitis (14). Предполагается, что оно может также иметь генетическую природу, но очень мало сообщений, описывающих это состояние у новорожденных (15-18). Анализ гена Tpit в трех таких случаях выявил две разные мутации в TPIT кодирующих последовательностях (11). После публикации этих оригинальных мутаций и распознавания клинической единицы, было идентифицировано больше случаев IAD у новорожденных и это привело к открытию многочисленных мутаций TPIT (13,19) и к первому описанию очень гомогенной клинической презентации неонатальной IAD (19). Мутации гена TPIT теперь оказались ассоциированными примерно с 2/3 IAD, проявляющихся у новорожденных (Рис. 2)б но никогда не наблюдалось ассоциации с IAD с ювенильным началом. Причина оставшихся 30% неонатальных IAD, не связанных с мутациями TPIT неясна, они могут быть связаны с мутациями регуляторных последовательностей TPIT, которые избегают идентификации и они могут включать мутации др. критических для дифференцировки кортикотрофов генов и/или для экспрессии РОМС. Неонатальные IAD чрезвычайно сходны с Tpit-/- мышами. Новорожденные с IAD обычно имеют не обнаружимые в плазме АСТН и кортизол и большинство их диагностируется вследствие тяжелых и внезапных гипогликемических эпизодов с судорогами приблизительно в трети случаев. В соответствии с этой тяжелой гипогликемией, обзор семейных историй болезни показывает, что многие носители в семьях имели необъяснимую гибель новорожденных, которая скорее всего обусловлена IAD. Один новорожденный был исследован при аутопсии в первую неделю после рождения. Выявлена двухсторонняя гипоплазия надпочечников и неспособность обнаружить с помощью иммуногистохимии АСТН, как и у Tpit-/- мышей (13).
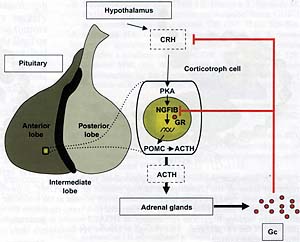
Fig. 3 Anterior pituitary corticotroph cells marate signals of the HPA axis. Hypothalamic CRH is the primary> stimulus of corticotroph cells: its action is exerted on release of pre-synthesized ACTH as well as on transcription of the POMC gene. The transcriptional effects of CRH signaling on POMC transcription are largely mediated through the orphan nuclear receptor NGFI-B. Negative feedback regulation of the HPA axis exerted by glucocorticoids (Gc) both at hypochalamic and pituitary levels. In pituitary, Gc inhibits ACTH release in parallel with repression of POMC gene transcription. HPA, hypothalamo-pituitary-adrenal; POMC. pro-opiomelanocortin.
Gc
Мутации TPIT идентифицированные у IAD пациентов включают нонсенсные, внутренние делеции и сплайсинг-мутации, которые скорее всего приводят к нонсенс-обусловленному распаду мРНК (21), и миссенс-мутации которые вызывают аминокислотные замены внутри Т домена. Большинство таких аминокислотных замещений вызывает потерю ДНК связывающей способности и как следствие потерю транскрипционной активности (11,13,19). Одна очень интересная мутация не предупреждает связывание с ДНК, но, по-видимому, специфически дефицитна по межбелковым взаимодействиям, включая гомодимеризацию Tpit, взаимодействие с Pitx1 и рекрутирование SRC2(IF2) ко-активатора (22). IAD обусловливается гомозиготной потерей функции TPIT от бессимптомных носителей родителей и поэтому способ передачи является рецессивным.
До идентификации
TPIT и его роли в функции кортикотрофов в гипофизе, IAD новорожденных распознавался с трудом как клиническая единица (23). Перевод регуляторных механизмов на клинический контекст явилось выразительной иллюстрацией новой клинической информации, предоставляемой базовой биологией.
Feedback regulation of the HPA axis
Первичным приводящим в движение НРА ось (Рис. 3) является гипоталямический corticotropin-releasing hormone (CRH), который секретируется в гипофизарную портальную систему в ответ на различные сигналы, включая циркадные ритмы и стрессы. Когда он достигает кортикотрофных клеток, то CRH связывается с 7 раз пронизывающим мембранным рецептором, который купирован с adenylate cyclase, приводя тем самым к активации пути протеин киназы А (РКА). Гипофизарные кортикотрофные клетки экспрессируют B-Raf так, что это делает возможным позитивное купирование пути РКА с путем mitogen-activated protein kinase и как следствие фармакологическая интерференция с каждым из этих путей вмешивается в ответ CRH (24,25).
У кортикотрофов передача сигналов CRH ведет к стимуляции высвобождения АСТН и к активации транскрипции РОМС. Активация транскрипции прежде всего обеспечивается посредством активации орфановых ядерных рецепторов, родственных NGFI-B/Nur77 (26). Lh/ члены этого подсемейства ядреных рецепторов включают Nurr1 и NOR1 (27). Хотя NGFI-B мРНК реагирует как непосредственно ранний отвечающий ген, очевидно, что NGFI-B белок всегда присутствует в клетках и что первым эффектом передачи сигналов CRH является дефосфорилирование NGFI-B DBD и гиперфосфорилирование его N-терминального AF1 домена (27). Это ведет к образованию димеров NGFI-B и рекрутированию этих димеров на чувствительный к Nur элемент гена РОМС (26,28). Димеры Nur фактора рекрутируют транскрипционный ко-активатор для своего действия и тот же самый транскрипционный ко-активатор, SRC2(TIF2), рекрутируется также на Trip в ответ на передачу сигналов CRH (25). Итак, эти измернения на промоторе РОМС ведут к усилению транскрипции. Амплитуда чувствительности к CRH, по-видимому, закладывается с помощью уровня Rb и RB-родственных белков в кортикотрофных клетках (29), а действие Rb обеспечивается посредством множественных белковых взаимодействий с NGFI-B и ко-активатором SRC2.
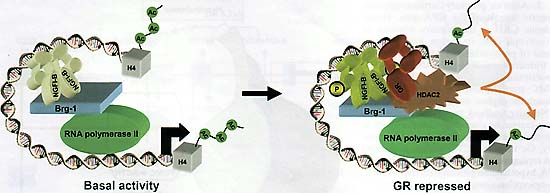
Fig. 4. A molecular mechanism for glucocorticoid receptor (GR)-mediated repression of the POMC gene. These diagrams represent some of the transcription factors present at the POMC promoter in basal condition, including NGFI-B. The chromatin remodeling ATPase Brgl is constitutively present at the promoter. Upon activation of GR by glucocorticoids, GR and the histone deacetylase HDAC2 are recruited to a trans-repression complex at the promoter; formation of this complex absolutely requires Brgl. The recruitment of HDAC2 results in deacetylation of histone H4 both within the promoter and the body of the POMC gene. This change in histone H4 acetylation likely contributes to transcriptional repression. POMC, pro-opiomelanocortin.
Гипофизарный АСТН высвобождается в ответ на CRH и быстро стимулирует синтез надпочечного Gc. Плазматический Gc образует негативную петлю обратной связи как для гипоталамуса, так и гипофиза, чтобы ингибировать синтез и высвобождение гипоталямического CRH и гипофизарного АСТН, соотв. Эта негативная петля обратной связи является критическим признаком НРА оси и потеря этой обратной петли имеет существенные патофизиологические последствия. Напр., хронический стресс может приводить к десенсибилизации Gc негативной обратной связи (30); также психиатрические заболевания, такие как шизофрения и депрессивные нарушения, ассоциируют с нарушением петли обратной связи Gc (31,32).
Molecular mechanisms of pituitary glucocorticoid feedback
Gc осуществляет свою негативную обратную связь на гипофизарные кортикотропины на двух уровнях параллельно; оба появляются очень быстро в течение минут и обеспечиваются посредством glucocorticoid receptor (GR)/C одной стороны, GR ведет к ингибированию высвобождения АСТН и параллельно репрессирует транскрипцию гена РОМС. Так, для активации транскрипции РОМС в ответ на CRH, имеет место Gc-зависимая репрессия на уровне инициации транскрипции. Разные механизмы были предложены для обеспечения GR репрессии РОМС: описан негативный к Gc чувствительный элемент, который обладает уникальным свойством связывать три молекулы GR (33) и показано, что он обеспечивает Gc репрессию в определенном геномном контексте (34). Gc, как было также показано, репрессирует транскрипцию РОМС благодаря антагонизму NGFI-B-зависимой транскрипции (35,36) В этом случае GR не связывается непосредственно с промотором РОМС, а скорее обеспечивает свой транскрипционный эффект посредством межбелковых взаимодействий с NGFI-B. Этот механизм известен как транс-репрессия и сходен с механизмом, описанным для GR репрессии АР1 или NFkB-зависимой транскрипции (37,38).
GR транс-репрессия NGFI-B-зависимой транскрипции, как установлено, зависит от ядерных белков, которые обеспечивают поддержку репрессивного комплекса с GR и NGFI-B на промоторах мишенях. В самом деле, АТФазная субъединица ремоделирующего хроматин Swi/Snif комплекса Brg1, по-видимому, существенна для стабилизации GR и NGFI-B взаимодействий в транс-репрессивном комплексе (39). Brg1 является также критическим для рекрутирования гистоновой деацетилазы HDAC2 на этот комплекс. Зависимость от Brg1 для транс-репрессии, по-видимому, является абсолютной, т.к. деплеция Brg1 из линии кортикотрофных модельных клеток устраняет способность GR репрессировать транскрипцию
РОМС и наоборот, GR/NGFI-B транс-репрессия может быть восстановлена в Brg1-дефицитных клетках в ответ на экспрессию Brg1 (39). Очевидно, что транс-репрессивный комплекс, формируемый на промоторе
РОМС, действует в основном посредством модуляции инициации транскрипции (Рис. 4). Транс-репрессия вкорее всего использует деацетилирование гистонов как промотора, так и гена с помощью HDAC2, т.к. уровень ацетилирования гистонов коррелирует с транскрипционной активностью (39).
Hormone resistence on Cushing's disease
Hypercortisolism или синдром Cushing имеет многочисленные метаболические последствия, которые могут в конечном итоге приводить к гипертензии, диабету и остеопорозу (40). Болезнь Кушинга является состоянием гиперкортисолемии, которое возникает из обычно небольшой злокачественной кортикотрофной аденомы гипофиза. Характерным признаком таких кортикотрофных аденом является их резистентность к Gc и их нечувствительность к обычной Gc негативной петле обратной связи. Потеря Gc обратной связи у гипофизарных кортикотрофов может быть ранним событием в туморогенезе кортикотрофов. Механизм Gc резистентности в кортикотрофных аденомах лишь иногда объясняется мутациями в GR (41).
Идентификация посредством молекулярных механистических исследований существенной роли Brg1 и HDAC2 в Пс репрессии гипофизарного РОМС позволяет предложить гипотезу, что потеря любого из этих белков может вызывать Gc резистентные аденомы (39). Эта гипотеза была оценена непосредственно на панели кортикотрофных аденом, полученный вследствие гипофизарной хирургии и было установлено, что около 50% этих опухолей дефицитны по ядерной экспрессии или Brg1 или HDAC2 (Рис.5). Т.к. некоторые опухоли теряли экспрессию любого из белков, то было сделано важное наблюдение на субнаборе аденом, в которых Brg1 всё ещё экспрессируется, но смещается в цитоплазму опухолевых клеток, тогда как она остаётся ядерной в окружающих здоровых клетках гипофиза (39). Безусловно, что группа опухолей, которые обнаруживают цитоплазматическую неправильную локализацию, обладает наиболее высокая пропорция пациентов, которые сохраняют частичную чувствительность к высоким дозам кортикоидов. Это может указывать на то, что первичный цитоплазматический Brg1 также присутствует на низких уровнях в ядрах этих опухолей. Эти наблюдения заставили расширить группу пациентов. Сходные наблюдения были сделаны на собаках с кортикотрофными аденомами, это подтверждает, что механизм Gc репрессии и Gc резистентности законсервирован, по крайней мере, у двух видов.
Эти исследования подтвердили молекулярный механизм патогенеза Gc резистентности при болезни Кушинга и подтвердили молекулярный механизм GR транс-репрессии гена
РОМС.

Fig. 5. Human corticotroph adenomas from Cushing's disease patients have aberrant expression of either Brgl or HDAC2. This table presents the analysis of 36 human corticotroph adenomas obtained at surgery for expression of Brgl and HDAC2. About 50% of these tumors presented with aberrant expression of either protein: expression defects included loss of expression in tumor cells (but not in surrounding healthy pituitary cells) or mis-localization Brgl to the cytoplasm in tumor cells (but not in surrounding healthy pituitary cells). Cushing's disease patients have been subdivided as resistant to low- (but not high) dose dexamethasone challenge as opposed to those patients who were also resistant to the high-dose dexamethasone suppression test.
Perspective
Помимо гормональной резистентности идентификация Brg1/HDAC2 как наиболее вероятного молекулярного виновника кортикотрофных аденом подтвердило также, что др. функции, контролируемые с помощью Brg1/HDAC2 могут вносить вклад в туморогенез кортикотрофов. В самом деле, Brg1, как полагают является опухолевым супрессором, а анализ экспрессии Brg1-зависимых генов в кортикотрофных аденомах уже подтвердил пути, которые могут участвовать в дерегуляции роста этих опухолевых клеток.
Сайт создан в системе
uCoz Bilodeau S., Vallette S.
Bilodeau S., Vallette S.