Diana E. Jaalouk & Jan Lammerding Nature Reviews Molecular Cell Biology 10, 63-73 (January 2009) | doi:10.1038/nrm2597
Cells sense their physical surroundings through mechanotransduction — that is, by translating mechanical forces and deformations into biochemical signals such as changes in intracellular calcium concentration or by activating diverse signalling pathways. In turn, these signals can adjust cellular and extracellular structure. This mechanosensitive feedback modulates cellular functions as diverse as migration, proliferation, differentiation and apoptosis, and is crucial for organ development and homeostasis. Consequently, defects in mechanotransduction — often caused by mutations or misregulation of proteins that disturb cellular or extracellular mechanics — are implicated in the development of various diseases, ranging from muscular dystrophies and cardiomyopathies to cancer progression and metastasis.
Martin, A. C. et al. Pulsed contractions of an actin–myosin network drive apical constriction. Nature 23 Nov 2008 (doi: 10.1038/nature07522)
Rauzi, M. et al. Nature and anisotropy of cortical forces orienting Drosophila tissue morphogenesis. Nature Cell Biol. 2 Nov 2008 (doi: 10.1038/ncb1798)
Во время гаструляции D. melanogaster апикальные сужения вентральных клеток облегчают образование вентральной борозды и последующей интернализации презумптивной мезодермы. Eric Wieschaus с коллегами исследовали, как миозин, локализующийся в апикальном кортексе суживающихся клеток вентральной борозды, создает силы для осуществления сужения. Они показали, что апикальное сужение клеток вентральной борозды является пульсовым - повторяющиеся сокращения, которые были асинхронными в соседних клетках, прерывались паузами, во время которых суженное состояние клеточных верхушек сохранялось. Миозиновые точки на апикальном кортексе были динамичными, они повторно увеличивались в своей интенсивности и перемещались совместно, чтобы сформировать более крупную и более интенсивную миозиновую структуру в медиальной части апикального кортекса. Эти взрывы скоплений миозина коррелировали с пульсами сжатия, это указывает на то, что сужения управляются с помощью контракций медиальной части апикального кортекса.
Транскрипционные факторы Twist и Snail дифференциально регулируют пульсовые контракции. В противоположность вентральным клеткам дикого типа, мутантны twist и snail накапливают миозин преимущественно в клеточных соединениях. Эксперименты с нокдауном показали, что экспрессия snail инициирует сокращения актин-миозиновой сети, в то время как экспрессия twist стабилизирует суженное состояние клеточной верхушки. Авт. предлагают 'ratchet' модель для апикального сужения, в которой фазы контракции актин-миозинового цитоскелета и стабилизации повторяются, чтобы сузить клеточную верхушку поэтапно.
Механические силы придают форму многим морфогенетическим изменениям во время развития D. melanogaster , включая элонгацию эмбрионов. Лежащий в основе механизм явился главной целью исследования Thomas Lecuit, Pierre-Franc,ois Lenne с коллегами, которые отслеживали индивидуальные клетки в целой ткани в течение времени и измеряли кортикальное натяжение во время элонгации зародышевого диска. Во время этого процесса ремоделирование поляризованных соединений, которое контролируется с помощью поляризованного обогащения myosin II, управляет обменами соседними клетками (интеркаляцией). Используя количественный анализ и математическое моделирование они показали, что анизотропия кортикального напряжения в апикальных клеточных соединениях достаточна для управления уменьшением объёма соединений, клеточной интеркаляции и элонгации ткани. Напряжение распределено анизотропно и зависит от накопления myosin II. Дальнейший анализ структур высшего порядка показал, что флюктуации в апикальном кортексе облегчают интеркаляции, это указывает на существование дополнительных сил, чтобы онги действовали на клеточные соединения.
Путем подсчета натяжения in vivo, может оказаться возможным определить, как внешние и внутренние силы кооперируют, чтобы управлять разнообразием форм и паттернов, наблюдаемых во время тканевого морфогенеза и определить, могут ли такие силы влиять обратно на др. онтогенетические процессы.
Механотрансдукция описывает клеточные процессы, которые транслируют механические стимулы в биохимические сигналы, тем самым позволяя клеткам адаптироваться к их физическому окружению. Известно несколько молекулярных игроков, которые участвуют в клеточной трансдукции (Box 1). Однако многие компоненты и особенно качественные особенности первичных механорецепторов остаются неполностью установленными.
Исследования механотрансдукции часто концентрируются на , таких как волосковые клетки внутреннего уха. Эти специализированные клетки часто участвуют в специфических клеточных структурах (Fig. 1), которые приспособлены к преобразованию механических стимулов в биохимические сигналы (напр., путем открытия ионных каналов в ответ на прилагаемые силы) и тем самым представляют собой прекрасную модель для изучения клеточной механочувствительности. Очевидно, что передача сигналов механотрансдукции играет критическую роль в подержании многих тканей, подвергающихся механическим воздействиям, такие как мышцы. кости, хрящи и кровеносные сосуды. Поэтому исследования проводятся на разных типах клеток, таких как миоциты, эндотелиальные клетки и , которые показывают, что механотрансдукция участвует в широком круге клеточных функций, не только субнаборе специализированных клеток и тканей. Напр., стволовые клетки могут детерминироваться в направлении специфических клеточных судеб на базе геометрии и жесткости субстрата, на котором клетки растут1 и на внутриклеточных физических взаимодействиях, таких как натяжение и адегзия, которые могут быть важны для эмбрионального развития как концентрационные градиенты морфогенетических факторов (see the Review by Wozniak and Chen88 in this issue).
Мы рассмотрим, как мутации и модификации, которые вмешиваются в нормальную механотрансдукцию и клеточную чувствительность к механическим стрессам, могут участвовать в широком спектре болезней в пределах от потери слуха до мышечной дистрофии и рака (Table 1). Многие из этих болезней обладают некоторым сходством. Однако, как мышечная дистрофия может быть связана с атеросклерозом или почечной недостаточностью? Их объединяют дефекты механотрансдукции, обусловливающие некоторое неожиданное сходство. Общим знаменателем многих механобиологических болезней является нарушение замысловатой передачи сил между внеклеточным матриксом (ECM), цитоскелетом и внутренностью ядра (Fig. 2). Клеточная механочувствительность базируется на индуцируемых силами конформационных изменениях в , которые являются предметом молекулярных сил, которые в результате открывают мембранные каналы или меняют сродство с партнером по связыванию, активируя тем самым сигнальные пути. Следовательно, любое изменение передачи обычных внутриклеточных сил посредством изменения в клеточной (или внеклеточной) структуре и организации может приводить к изменению молекулярных сил, действующих на эти белки, приводящих к ослаблению или усилению механочувствительных сигналов. Помимо дефектов, которые затрагивают клеточную структуру и организацию, и тем самым клеточную механочувствительность, мутации в белках, которые участвуют в нижестоящих путях передачи сигналов, также могут вызывать нарушения механотрансдукции. Примеры включают мутации белков, которые участвуют во внутриклеточной передаче сигналов кальция, или членов Rho или mitogen-activated protein kinase (MAPK) пути2. В общем любые изменения в клеточной или внеклеточной структуре, клеточных механочувствительных процессах сами по себе или последующие нижестоящие сигнальные пути могут приводить к изменению или аномалиям механотрансдукции и приводить к болезням (Fig. 3). Идентификация молекулярных деталей, которые участвуют в нормальной и дефектной механотрансдукции д. привести к лучшему пониманию механизмов. лежащих в основе болезней и нормальной клеточной функции.
Cells need to feel the force
Все клетки и организмы всего эволюционного спектра от наиболее примитивных до наиболее сложных являются чувствительными к механическим воздействиям3, 4. Это 'универсальное' свойство позволяет клеткам воспринимать механические стимулы от своего физического окружения или от имеющихся в организме электрохимических или биохимических сигналов, которые затем регулируют широкий репертуар физиологических реакций. Как таковая механотрансдукция - участвует в перцепции внешне приложенных сил (напр., ощущение прикосновения) или в регуляции сил и напряжений в теле (напр., в мышечном натяжении или регуляции кровяного давления) - является важной для путей развития и для нормального гомеостаза тканей, в первую очередь для поддержания тканей, в которых клеточные адаптивные реакции являются критическими для противодействия существенным вариациям нормальных условий4, 5. Напр., мышечная ткань отвечает на упражнения гипертрофическим ростом - т.е. имеет место увеличение размера клеток. Сосудистая система может поддерживать постоянное кровяное давление несмотря на изменения в кардиальных выбросов за счет сужения или расширения сосудов (контракции или реляксации гладкомышечных клеток, которые окружают кровеносные сосуды).
Один из наиболее часто цитируемых примеров механотрансдукции является её роль для слуха и баланса, которые вытекают из электрохимический реакций на звуковые волны. давление и тяжесть (see Review by Chalfie89 in this issue). Эти механические силы вызывают незначительные смещения в волосковых клеток внутреннего уха. Отклонение стереоцилий вызывает натяжение кончиковых связок, небольших внеклеточных филамент, которые соединяют кончики стереоцилий, тем самым происходит натяжение механически открывающее ионные каналы (Fig. 1). Быстрый приток кальция и др. ионов может затем инициировать дальнейшую нижестоящую передачу сигналов. , которые располагаются на дистальных концах кончиковых связок могут расслабляться или сокращаться, чтобы восстановить остаточное натяжение, такой механизм позволяет системе адаптироваться к динамическим стимулам 6. Сходным образом механотрансдукция является жизненно важной для тактильной чувствительности и проприоце6пции (внутреннее ощущение относительного положения частей тела), которые обладают сходными подлежащими механизмами передачи сигналов механотрансдукции, как и при слухе7, 8.
Механотрансдукция также играет фундаментальную роль в регуляции физиологического феномена в др. специализированных тканях, которые непосредственно не участвуют в сенсорной функции. Напр., скелетные и кардиальные мышцы могут отвечать на повышенную нагрузку, на интенсивные упражнения гипертрофическим ростом, в то время как лишенные подвижности мышцы будут со временем атрофироваться9. Роль регуляторной механотрансдукции в сердечно-сосудистой системе особенно поразительна. Установлено, что морфология и физиология сердца и сосудистой системы испытывает влияние со стороны давления и касательных (shear) стрессов, которые генерируются кровотоком10-13, а низкая интерстициальная скорость тока достаточна для стимуляции лимфангиогенеза14. Исследования in vivo у эмбрионов рыбок данио показали, что иные high-shear паттерны тока присутствуют во время критических стадий развития сердца: искусственное нарушение shear усилий за счет закупорки приводит к аномальному образованию сердца (напр., аномальной третьей камере, уменьшению петлеобразования или дефектным клапанам) с дефектами. которые сходны с теми, что наблюдаются при некоторых врожденных пороках сердца15. В зрелой сердечно-сосудистой системе, как полагают, laminar shear стрессы и растягивание сосудов в диаметре вызывают атерозащитный эффект на эндотелиальные клетки. В соответствии с этой идеей атеросклеротические повреждения часто обнаруживаются в специфических местах нарушенного (т.е. турбулентного) характера тока, характеризующегося низкими и оссциллирующими shear стрессами на эндотелий (see the Review by Hahn and Schwartz90 in this issue).
Др. примером роли механотрансдукции в поддержании ткани является кость. Компактная кость представлена концентрическими слоями косного матрикса, в котором небольшие полости, известные как лакуны, вкраплены с регулярными интервалами. Эти лакуны содержат остеоциты, соединенные посредством сети взаимосвязанных канальцев. Тяжесть и сдавливающие силы, которые генерируются мышечными сокращениями во время движения приводят к небольшим деформациям poro-elastic кости, создавая градиенты давления, которые управляют интерстициальным током жидкости через lacunae-canalicular сеть. Этот индуцированный нагрузкой ток жидкости, как полагают, стимулирует локальное ремоделирование кости и оптимизирует физическую работу кости посредством сигналов механотрансдукции16. Сходным образом, хондроциты (основные клетки, которые представляют хрящ) адаптируются к широко варьирующим воздействиям за счет секреции богатого glycosaminoglycan ECM. который придает хрящу динамические механические свойства. Более того, легочная физиология от развития до созревания испытывает влияние со стороны постоянно изменяющихся механических воздействий и натяжений, которые вызываются циклическими растяжениями и контракциями легких17. Сходным образом, ток мочи внутрь почечных канальцев играет центральную роль в регуляции морфогенеза почек, также эти клетки ощущают shear stress жидкости за счет изгибания первичных18.
Mechanotransduction and disease
Способность клеток отвечать на изменения в их физическом окружении является критической для развития и поддержания тканей, которые испытывают различные механические воздействия (напр., мышцы и кости), а также для физиологических процессов, которые затрагивают весь организм (напр., контроль кровяного давления и кровотока). На клеточном уровне механотрансдукция может модулировать разнообразные функции, такие как белковый синтез, секрецию, адгезию. миграцию, пролиферацию, жизнеспособность и апоптоз. Соотв. дефекты клеточной механотрансдукции - часто посредством врожденных или приобретенных мутаций - могут приводить или могут , по крайней мере, вносить вклад в различные заболевания у людей (Table 1). Альтернативно, изменения в клеточном физическом окружении могут вызывать патологические последствия, даже когда процессы клеточной механотрансдукции функционируют должным образом. Примеры этого сценария включают нарушения fluid shear стрессов на бифуркациях. которые запускают сосудистое ремоделирование, которое может приводить к развитию атеросклероза19 или потере костной массы в условиях микрогравитации20. В этих случаях имеют место аномальные механические стрессы на клеточном уровне, которые - посредством (обычной) передачи сигналов механотрансдукции - модулируют клеточные процессы, которые могут приводить к нарушению нормальной функции ткани.
Т.к. почти все клетки используют передачу сигналов механотрансдукции для нормальной функции, то многие ткани могут характеризоваться нарушенной биомеханикой или механочувствительностью. Прекрасный пример - потеря слуха, которая вызывается мутациями в , которые кодируют механочувствительные белки6 (Fig. 1). Др. примеры затронутых тканей включают кость16, 20, хрящ, легкие21-23, иммунную систему24-26 и ЦНС27, 28. Рассмотрим дефекты механотрансдукции в скелетных и кардиальных мышцах, которые вызывают мышечную дистрофию или кардиомиопатии29-31 и коротко дефекты глаз.
Cardiac mechanosensing and hypertrophy. Более 400 различных мутаций было идентифицировано у пациентов с кардиомиопатией, затрагивающие 9 отдельных саркомерных генов, включая actin, α-tropomyosin, troponin, titin и наиболее часто, β-myosin heavy chain32. Чтобы понять. как мутации этих структурных белков могут приводить к патологической гипертрофии, рассмотрим эти белки в свете кардиальной механотрансдукции.
Кардиальные миоциты могут отвечать непосредственно на механическую деформацию или натяжение посредством нескольких внутренних механосенсоров, хотя точные механизмы механочувствительности остаются до конца не выясненными. Презумптивные механосенсоры включают клеточных мембран, integrins и integrin-ассоциированные белки (такие как melusin или integrin-linked kinase (ILK)), саркомерные белки (такие как titin, myosin или small LIM-domain protein MLP) и рецепторы клеточной поверхности (такие как G-protein-coupled receptors (GPCRs) и angiotensin II type 1 receptors), которые могут быть активированы с помощью событий натяжения даже в отсутствие лигандов. Эти механосенсоры активируют множественные и перекрывающиеся клеточные сигнальные пути, которые включают передачу сигналов Ras/Rho и MAPK, активацию phospholipase C, calcium/calcineurin-опосредованную передачу сигналов и microRNAs32 (Fig. 4). Эти пути запускают экспрессию гипертрофических генов и вызывают увеличение длины миоцитов и/или их толщину (reviewed in Refs 32, 33). Эти пути механотрансдукции вместе с часто перекрывающимися нейрогормональными механизмами (напр., передача сигналов GPCR , которая активируется с помощью angiotensin или catecholamines) позволяют сердцу адаптироваться к продолжительным изменениям в механических рабочих нагрузках за счет увеличения размеров кардиальных миоцитов (гипертрофии) и модификации окружающего ECM, обозначаемых как кардиальное ремоделирование.
Реакция кардиальной гипертрофии часто распадается на физиологическую и патологическую гипертрофию. Физиологическая гипертрофия, которая возникает как следствие аэробных упражнений или беременности, характеризуется последовательным добавлением (удлиняя клетки) и параллельно (увеличивая толщину клеток), приводя к увеличению толщины стенки сердца, а размеры камер приспосабливаются к повышенной гемодинамической нагрузке. Напротив, патологическая гипертрофия вызывается за счет аномальных изменений кардиальных рабочих нагрузок, напр., посредством гипертензии, , инфаркта миокарда или врожденных дефектов, которые обусловлены мутациями генов, которые кодируют саркомерные белки. Так, при физиологической гипертрофии кардиальные миоциты ощущают повышенное воздействие на стенки желудочков и отвечают увеличением размеров клеток. Однако при патологической гипертрофии миоциты часто обнаруживают диспропорциональное увеличение или в толщине, приводя к увеличению толщины стенки желудочков, или длины, приводя к расширению желудочков. Гипертрофическая реакция первоначально благоприятна, т.к. нормализует и поэтому часто обозначается как компенсаторная гипертрофия9, 34. Однако повышенные уровни стрессов, которые сохраняются в течение длительных периодов времени. часто ведут к неадекватному ремоделированию миоцитов и ECM, это сопровождается апоптозом и некрозом миоцитов и в конечном итоге приводит к сердечной недостаточности32.
Точные молекулярные механизмы, которые управляют переходом от компенсаторной гипертрофии к патологическому ремоделированию, остаются до конца не выясненными. Несколько линий доказательств подтверждают, что гипертрофия может ещё больше дестабилизировать кардиальную механику, т.к. гипертрофическая ткань часто характеризуется нарушением динамики сокращений и расслаблений. Помимо этого существует клеточная программа, которая ответственна за патологическую гипертрофию в результате повторной экспрессии генов, обычно ассоциированной с эмбриональным миокардом. Это вызывает дезорганизацию клеточной структуры, нарушение динамики кальция и усиленный интерстициальный фиброз, усиление механического дисбаланса между кардиальной функцией и 35. Неадекватное ремоделирование ECM также может приводить к к относительному смещению кардиальных миоцитов, усиливающем в дальнейшем механический дисбаланс в миокарде. Обнадеживающими находками у животных моделей являются то, что патологическая гипертрофия может быть предупреждена или даже устранена за счет модулирования сигнальных путе, которые участвуют в гипертрофической реакции, это заставляет искать специфические фармакологические модуляторы32, 33. Задачей в разработке этих терапевтических подходов является распутывание дихотомии физиологической и компенсаторной гипертрофии, с одной стороны, и патологической гипертрофии, с др. стороны, т.к. существует значительное перекрывание между сигнальными путями, которые вовлечены в эти процессы.
Mechanotransduction and muscular dystrophies. В клетках скелетных мышц силы, которые генерируются в саркомерах, передаются ECM посредством специализированных белковых комплексов, которые состоят из dystrophin и ассоциированных с dystrophin белковых комплексов а плазматической мембране (Fig. 2), тем самым клеточная мембрана защищается от избыточных воздействий. При мутации в гене dystrophin нарушают передачу сил между цитоскелетом и ECM, приводя к прогрессирующей мышечной дегенерации36. Важно, что разрушение связей цитоскелет-ECM не только наделяет клетки большей чувствительностью к повреждениям мембран, но и также вызывает аберрантную активацию передачи сигналов MAPK extracellular signal-regulated kinase 1 () и в ответ на растяжение37. Такая аномальная механотрансдукция может в дальнейшем воздействовать на функцию и жизнеспособность клеток. Недавние эксперименты показали, что dystrophin-дефицитные мышечные волокна в результате стрессами индуцированных разрывов более ломкой плазматической мембраны делаются способными притягивать внеклеточный кальций. Это вызывает аномальные мышечные сокращения и в комбинации с дефектными связями между цитоскелетом и ECM ведет к физическим повреждениям цитоскелета, которые впоследствие приводят к потере мышечных клеток38. Интересно, что дистрофин также участвует в реакции эндотелиальных клеток, которая необходима для расширений артерий, вызываемых fluid shear стрессами. Поэтому эндотелиальные клетки от dystrophin-дефицитных мышей обнаруживают нарушенную передачу сигналов механотрансдукции в ответ на fluid shear стрессы, приводя к истощающей дилятации артерий и к пониженной сосудистой плотности в кардиальной мышце, это может в дальнейшем приводить к прогрессирующей потере мышц39.
Сходным образом мутации в цитоскелетных белках desmin, titin и myosin, которые являются важными компонентами саркомеров, приводят к дезорганизации саркомеров и нарушению клеточной механики, включая нарушение генерации сил и нарушенную (пассивную) жесткость, которые могут нарушать динамику реляксации миоцитов. Повреждающие эффекты этих мутаций могут возникать за счет непосредственных изменений в распределении внутриклеточных сил и/или их генерации, обусловленной ультрастуктурной дезорганизацией, но ммогут также возникать и в результате нижестоящих эффектов изменения клеточной механочувствительности, т.к. myosin и titin могут функционировать как механосенсоры40. Изучение относительных вкладов в эти механизмы мышечной дисфункции могут дать важную клиническую информацию, т.к. дефекты путей механотрансдукции могут быть потенциально ослаблены фармакологическими реагентами. Эти исследования затруднены фактически из-за того. что механотрансдукция может непосредственно влиять на клеточную структуру и функцию, затрудняя вычленять причины и эффекты.
Недавние находки, что мышечные дистрофии могут возникать в результате мутаций белков ядерной оболочки (а именно lamins A и C, или nesprins) ещё сильнее подчеркивает концепцию, что нарушенные внутриклеточная структура и передача сил могут вносить вклад в мышечные болезни. Хотя эти белки экспрессируются в наиболее дифференцированных клетках возникающие фенотипы часто специфичны для мышц и указывают на то, что клетки у затронутых пациентов обладают повышенной чувствительностью к механическим стрессам. Новая информация получена на мышиной модели с отсутствием lamins A и C. Клетки таких животных характеризуются пониженной жесткостью ядер, повышенной ломкостью ядер и нарушенной активацией механочувствительных генов, вызывающих снижение жизнеспособности клеток, подвергающихся повторяющимся растяжениям41. Повышенная ломкость ядер д. приводить к разрывам ядер и клеточной гибели в тканях, подвергающихся механическим стрессам. Однако разрывы ядер могут объяснить лишь часть клеточной гибели, которая обнаруживается во время воздействия повторяющихся натяжений, особенно в клетках emerin-null мышей, которые имеют обычную ядерную механику и повышенный натяжением индуцируемый апоптоз42. Следовательно, очень возможно, что ослабление экспрессии механочувствительных анти-апоптических генов, таких как , вносит вклад в повышенную чувствительность клеток к механическим стрессам41, 42. Сегодня остается неизвестным, какова причина дефектов механотрансдукции в этих клетках. Хотя ядро часто считается клеточным механосенсором - напр., путем изменения доступности хроматина в ответ на деформации - прямые доказательства этой функции слишком зыбки и лишь будущие исследования смогут установить, возникают ли дефекты механотрансдукции как прямое следствие нарушения ядерной жесткости или это в основном отражает широкие дефекты в специфических сигнальных путях (напр., в передаче сигналов ядерного factor-kappaB) , которые модулируются ядерными lamins.
Trouble in the eye. Др. неожиданным органом. который затрагивается нарушением механотрансдукции является глаз. Имеющиеся данные указывают на то, что модулированные сигналы механотрансдукции, обусловленные повышенными механическими стрессами, могут существенно влиять на патогенез и 43. При глаукоме повышенное гидростатическое давление и измененная биомеханика зрительного нерва могут инициировать механизмы, которые ведут к потере зрения. Недавние экспериментальные доказательства подтвердили, что человеческие (и обезьяньи) глазные ткани деформируются в ответ даже на небольшие изменения в 44. Кроме того, повышение обтекающего гидростатического давления, напоминающее внутриглазные условия при глаукоме, может индуцировать апоптоз клеток ретинальных ганглиев in vitro, это согласуется с находками in vivo44. Более того, при фиброзе склер у людей (первичных клеток, которые участвуют в ремоделировании склер, которое сопровождает аксиальное удлинение во время развития миопии) экспрессируются многие гены, которые модулируются воздействиями механических натяжений. Сюда входят гены, которые кодируют белки ECM (такие как ), протеин киназы (такие как lymphocyte-specific protein tyrosine kinase (LCK) человека), клеточные рецепторы (такие как parathyroid hormone (PTH)/PTH-related peptide (PTHrP) receptor), факторы клеточного роста и дифференцировки (такие как fibroblast growth factors and bone morphogenetic proteins) и транскрипционные факторы (такие как Jun B)45. Хотя роль некоторых из этих белков в развитии глаз и осевой элонгации неясна и довольно спекулятивна, вклад др. более очевиден. Напр., активация PTH/PTHrP рецептора с помощью гормонов. регулируемых кальцием, запускает несколько внутриклеточных сигнальных событий, включая активацию protein kinase C, которая непосредственно участвует в ремоделировании склер. Более того, tenascin C, который участвует в тканевом ремоделировании во время развития может действовать как медиатор реакций склер на натяжение, повышающее синтез протеолитических энзимов, которые влияют на ремоделирование ECM. Эти находки указывают на то, что повышенное внутриглазное давление - обеспечиваемое с помощью обычной передачи сигналов механотрансдукции в фибробластах склер - может вносить вклад в аномальное ремоделирование, которое происходит при аксиальной миопии.
Development and premature ageing
Большое количество доказательств указывает на то, что механотрансдукция играет также критическую роль в развитии46-49. Т.о., любые нарушения механизмов обычной механотрансдукции или клеточных физических условий могут приводить к различным дефектам развития50. Классическим примером, иллюстрирующим это является , который характеризуется обратным лево-правосторонним расположением первичных висцеральных органов. Формирование лево-правостороннего паттерна у ранних эмбрионов млекопитающих диктуется управляемым ресничками лево-направленным током жидкости во время беременности, который - посредством передачи сигналов механотрансдукции - дифференциально индуцирует экспрессию nodal, молекулы семейства transforming growth factor (TGF), и сигнальный каскад др. факторов на левой стороне эмбриона. Мутации в dynein моторных белках, которые в первую очередь отвечают за синдром Kartagener's, блокируют движения ресничек в эпителии узелка срединной линии у эмбриона и тем самым предупреждают лево-направленный ток жидкости; в отсутствие тока формирование лево-правостороннего паттерна становится случайным.
Сходным образом, мутации в генах , которые кодируют белки ресничек polycystin 1 или transient receptor potential channel family protein polycystin 2 (TRPP2) предоставляют прямые доказательства дефектов механотрансдукции, которые ведут к болезни почек51. Polycystins формируют чувствительные к механическим воздействиям ионные каналы в ресничках почечных эпителиальных клеток и делают возможным приток кальция в ответ на индуцируемое током жидкости изгибание ресничек, действуя как клеточные сенсоры тока в почках51. Polycystin мутации, которые ассоциируют с потерей функции делают клетки неспособными ощущать ток жидкости, который обычно регулирует морфогенез почек, приводя к тяжелыми типам polycystic kidney disease (PKD), которые включают аутосомно доминантную PKD, характеризующуюся прогрессивным образование кист, которые достигают кульминации при деструкции почек51, 52.
Нарушение механотрансдукции может также лежать в основе некоторых др. болезней, которые обычно не рассматриваются в биофизической перспективе. Одним из таких примеров является (HGPS), прогероидное нарушение, которое вызывается мутациями в гене LMNA, кодирующем lamin A. Пациенты с HGPS выглядят нормальными при рождении, но не способны процветать вскоре после этого и погибают в возрасте от 13 до 19 лет. Атеросклероз является основной причиной, ведущей к гибели пациентов с HGPS53, а post-mortem анализ сосудистых тканей от пациентов с HGPS и от мышей, моделирующих HGPS, выявил интенсивную потерю сосудистых гладкомышечных клеток и необычную чувствительность к гемодинамическим стрессам54, 55. Механотрансдукция в сосудистых клетках в ответ на fluid shear стрессы и растяжения из-за увеличения сосудов является критическим защитным механизмом против атеросклероза и может обеспечиваться апоптозом, пролиферацией и секрецией ECM в здоровых сосудистых гладкомышечных клетках56. Недавние эксперименты в нашей лаб. показали, что фибробласты от пациентов с HGPS обнаруживают пониженную жизнеспособность, если подвергаются повторяющимся механическим натяжениям и что клетки от пациентов с HGPS лишены вызываемой натяжением реакции пролиферации, которая обычно наблюдается в клетках от здоровых людей57. Эти находки указывают на то. что повышенная клеточная чувствительность сосудистых клеток. подвергается обычным fluid shear стрессам и расширение сосудов может быть возможным механизмом для прогрессивной потери гладкомышечных клеток и тяжелого атеросклероза у пациентов с HGPS. Хотя повышенная клеточная чувствительность к механическим натяжениям определенно не единственный фактор при HGPS, наши эксперименты подтверждают, что он мможет быть важным для развития тяжелого атеросклероза, который ведет к летальным инсультам и инфарктам миокарда у пациентов55. Более того, он вносит вклад в дефекты др. тканей, подвергающихся механическим нагрузкам, вообще-то вызывает аномалии костей и дистрофию скелетных мышц, которые характерны для HGPS.
Cancer cells have lost their touch
Наиболее интригующим изо всех болезней механотрансдукции является рак. Внезапные изменения в ECM механике, ECM ремоделировании и возникающих в результате нарушений цитоскелетного натяжения и передачи сигналов механотрансдукции выявляются как важные факторы, которые могут способствовать злокачественному перерождению, туморогенезу и метастазам58-60. В дополнение к комбинации генетических мутаций и повышенной активности онкогенов, реорганизации цитоскелета - особенно те, которые проявляются в виде альтераций сил натяжения, которые генерируются actin-myosin аппаратом в клетке - играют жизненно важную роль в морфологических изменениях, которые адаптируются опухолевыми клетками, т.к. они приобретают инвазивный фенотип. Одним из основных регуляторов натяжения цитоскелета являются Rho семейство GTPases. Среди их многочисленных мишеней, Rho действует посредством Rho kinase (ROCK), чтобы регулировать фосфорилирование легкой цепи миозина, посредством ингибирующего фосфорилирования myosin phosphatase. Хотя исследования активности Rho в опухолях приводят к противоречивым результатам (некоторые сообщения предоставляют доказательства, подтверждающие мнение, что опухоли обладают повышенной Rho активностью и обнаруживают боле высокое натяжение цитоскелета, тогда как др. сообщают о снижении активности Rho в солидных опухолях61-66) становится очевидным, что натяжение цитоскелета существенно влияет на сигнальные пути, которые участвуют в прогрессировании рака. Расхождения этих исследований могут быть частично приписаны разным экспериментальным условиям, которые используются и ограничениям, которые ассоциируют с двумерными (2D) монослойными культурами клеток. в
Более того, некоторые исследования показали, что натяжение цитоскелета в опухолях зависит от жесткости ECM2, 58, 67. Опухоли обычно намного тверже, чем окружающая нормальная ткань. Конкурентные изменения в жесткости ткани, опухолевый рост, обусловленный пролиферирующими клетками, и/или повышенное давление интерстициальной жидкости все он комбинируются, чтобы повлиять на физическое окружение раковых клеток внутри опухоли и в соседних нормальных клетках68. Такое измененное физическое окружение может модулировать судьбы этих клеток посредством механотрансдукции (Fig. 5). Напр., высокая жесткость ECM может приводить к нарушению нормальной полярности эпителиальных клеток, заставляя эпителиальные клетки млекопитающих заполнять просветы кист при раке груди2. Paszek с сотр. исследовали, может ли повышенная жесткость ткани, которая наблюдается в опухолях молочных желез, в 3D матрицах способствовать злокачественному фенотипу путем воздействия на integrins, рецепторы клеточной поверхности, которые соединяют специфические молекулы ECM с цитоскелетом2 (Fig. 2). Они установили. что жесткость матрикса (экзогенная сила) и натяжение цитоскелета (эндогенная сила) функционально кооперируют с 'mechano-circuit', который модулирует фенотипические трансформации в опухолях, путем купирования механочувствительной роли интегринов в передаче внешних физических стимулов с сигнальными путями Rho и ERK. Как таковая жесткость матрикса нарушает эпителиальный морфогенез, вызывая зависимую от сил агрегацию и образование кластеров интегринов, приводя тем самым к повышению Rho-ROCK-зависимого цитоскелетного натяжения, которое умножает формирование и стабилизацию . Такое усиление клетками продуцируемой силы и сборки фокальных адгезий сопровождается передачей сигналов с помощью focal adhesion kinase, ROCK-обусловленным нарушением слипчивых соединений, усилением зависимой от фактора роста активации ERK, управляющей пролиферацией опухолевых клеток и нарушением базальной полярности, тем самым устраняя образование просветов и ремоделируя архитектуру ткани молочных желез. Нарушение передачи сигналов Rho или ERK , чтобы редуцировать натяжение цитоскелета до нормального уровня приводит к существенному снижению пролиферации опухолевых клеток и к репрессии злокачественного фенотипа. Недавние эксперименты продемонстрировали также, что как интегринами, так и Rho-обеспечиваемая регуляция внутриклеточного натяжения необходимы для обеспечения инвазивного поведения фибробластов и раковых клеток в ко-культурах69, 70.
Все клетки за исключением гематопоэтических клеток, нуждаются в прикреплении к твердому субстрату для нормального хода клеточного цикла и жизнеспособности. Так, раковые клетки теряют свою зависимость от прикрепления и натяжения клеточной поверхности, т.к. приобретают способность проникать в др. ткани71, 72. Это характерный для метастатических клеток признак, характеризует из способность прорываться через базальную ламину, проникать в кровеносные сосуды, выходить из кровеносных сосудов и формировать новые опухоли, он нуждается в токо регулируемых взаимодействиях между раковыми клетками и их физическим окружением. Напр., адгезия меланомных клеток с эндотелиальными клетками, которые выстилают кровеносные сосуды (критическая ступень экстравазации и метастазирования) частично регулируется величиной гидродинамического касательного напряжения (shear), которое обеспечивает агрегацию меланома-лейкоцит за счет усиления адгезии опухолевых клеток с эндотелием73. Более того, хотя опухоли тверже, но метастатические клетки могут отличаться от клеток неинвазивного рака и от нормальных клеток снижением жесткости цитоскелета и повышенной деформируемостью60, 74, 75. Недавние доказательства подтверждают, что способность клеток к деформациям строго коррелирует с временем прохождения через узкие поры и с повышенным метастатическим потенциалом клеток мышиной меланомы76. Т.о., повышенная клеточная и ядерная деформируемость может способствовать прохождению метастатических раковых клеток через поры ограниченного размера и кровеносные сосуды, приводя к усилению метастазирования. Хотя некоторые из этих исследований, чтобы измерить жесткость раковых клеток, сталкиваются с техническими ограничениями (напр., обычно измерения слипчивости клеток осуществлялись в суспензиях или лишь с небольшим количеством тестируемых клеток), эксперименты иллюстрируют, что многие раковые клетки характеризуются измененными физическими свойствами и что биомеханические измерения клеток изолированных из могут иметь важное диагностическое и прогностическое значение.
Ясно, что рак вызывается не только дефектной передачей сигналов механотрансдукции, т.к. дерегуляция контроля клеточных циклов, дефекты репарации ДНК, супрессия апоптоза и изменение адгезии и/или миграции все это вносит вклад в это болезнь со многими лицами. Однако многие из этих клеточных функций, которые вовлечены в туморогенез и метастазирование модулируются с помощью механотрансдукции. Следовательно, изменения механотрансдукции могут быть важными компонентами формирования опухолей и метастазирования. Улучшенные условия клеточных культур, которые позволять изучать прогресс опухолей в 3D контексте, существенно повысят нашу способность к расшифровке эффектов нарушений механотрансдукции при прогрессировании опухолей.
Conclusions and future perspectives
The above examples demonstrate that the mechanotransduction feedback loops that couple cellular structure and function, and that modulate cellular structure and the extracellular environment, play an important role in the maintenance of normal tissue function. Moreover, events that disrupt these feedback loops, either by affecting cellular mechanosensing, intracellular mechanotransduction signalling or intracellular or extracellular force distribution, can result in various clinical phenotypes.
One challenge that remains is to determine the effects of defective structural or motor proteins on mechanotransduction. Often, structure, function and mechanotransduction are tightly linked, as the examples of myosin, titin (both recently identified as potential cellular mechanosensors), talin and lamins illustrate. Talin and vinculin are structural components of the focal adhesion complex, linking integrins to actin filaments (Fig. 2). Molecular dynamic models suggest that force-induced conformational changes in talin can activate a cryptic vinculin binding site, enabling subsequent recruitment of vinculin to reinforce focal adhesion sites77. Similarly, lamins were first identified as nuclear intermediate filaments, but were subsequently shown to have an important role in transcriptional regulation as well as in DNA and RNA synthesis78. These examples highlight our still limited understanding of the cellular structure–function relationships — that is, we still do not fully understand how the 3D organization of the cytoskeleton and the nucleus affect cellular functions such as DNA and RNA transcription. Future research should focus on how changes in intracellular structure, through induced deformations or remodelling for example, can modulate these cellular functions and processes.
Understanding these processes may also provide us with new clues in the search for the elusive cellular mechanosensors and the question of how cells manage to sense their physical surroundings. The many reports of putative mechanosensing proteins suggest that multiple mechanisms exist, even in single cell types, so that the interplay of redundant or complementary mechanotransduction pathways has to be viewed in a 'systems biology' context3. This will be especially important when designing treatment approaches for mechanotransduction diseases. Although mutations in structural proteins may require the replacement of affected genes or cells by targeted gene or stem-cell therapy — currently a challenging and daunting task — an alternative approach for at least some of these diseases could be to correct downstream signalling pathways that are disturbed by altered mechanotransduction signalling. For some dominant-negative mutations, another strategy could be based on the possible redundancy of related proteins; it might be possible to reduce levels of the mutant protein using RNA interference. Such an approach has been proposed for a lamin A mutation that is associated with HGPS, as studies in a lamin A-deficient mouse model suggest that lamin C might be sufficient to maintain cellular function without an increase in cellular sensitivity79. Alternatively, some cellular mechanical defects can potentially be directly addressed using small molecules approaches. For example, the membrane sealant poloxamer 188 has been successfully used to reduce damage to the plasma membrane in a mouse model of Duchenne muscular dystrophy, resulting in significant improvement in cellular function in cardiac and skeletal muscle80, 81.
The past years have provided increasing evidence that the finely tuned feedback between cells and their physical surroundings is crucial for many important cellular functions, ranging from differentiation to proliferation and viability. Events that interfere with these cellular mechanotransduction processes may thus result in diseases that affect various tissues and organs. Studying the mechanisms that underlie these diseases may lead us to new treatment strategies, improved tissue-engineering design and enhanced biomaterials, and will also provide us with an opportunity to learn more about mechanotransduction and mechanobiology in normal cells and physiology.
The Force Is with Us Martin A. Schwartz Science Vol. 323, P.588-589, 2009
The ability of cells and tissues to respond to mechanical force is central to many aspects of biology. Areas relevant to human physiology and disease include development and maintenance of bone, blood vessels, and muscles; regulation of blood pressure; motility of cells; and
regulation of cell proliferation (1). Mechanosensitive adhesions mediated
by membrane proteins called integrins are believed to underlie a number of
these behaviors (2). Forces applied either by the cells' own cytoskeleton or from external sources induce strengthening of these adhesions and trigger a variety of intracellular signals through these effects. Two papers in this issue, one by Friedland et al. on page 642 (3) and the other by del Rio et al. on page 638 (4),increase our understanding of the molecular basis for these phenomena.
Current models for mechanotransduction generally involve alterations in
protein conformation by forces (1, 5). Stretch-activated membrane channels,
present in cells from all organisms that have been examined, undergo conformational transitions in response to changes in membrane tension. For cytoskeletal and extracellular matrix proteins that form linear polymers, tension is thought to cause partial or complete unfolding of domains, exposing new binding sites orother motifs. The extracellular matrix
protein fibronectin is the best-studied example. Fibronectin requires tension to assemble into fibrils (6, 7), and fluorescence resonance energy transfer measurements showed that fibronectin in fibrils is in an extended state (8). Stretching fibronectin exposes a cryptic binding site
in the first type III domain, leading to associations between fibronectin proteins that promote fibril formation (6).
Integrins are one link in a connection that runs from extracellular matrix proteins such as fibronectin across the plasma membrane to the actin cytoskeleton. Integrins form this connection in part by binding the cytoskeletal protein talin, which binds actin directly, as well as by recruiting the cytoskeletal protein vinculin, which also binds
actin (9). Because every component in this chain experiences tension, each one is in principle a potential mechanotransducer.
The best-defined response of integrindependent cell adhesions to tension is reinforcement of the adhesions to resist the applied
force (2). These responses occur over several time scales. Over tens of seconds, vinculin is recruited to small adhesions, most likely without recruitment of additional integrins (10). Over a few minutes, adhesions grow
larger and lengthen in the direction of the applied force (11), which appears to involve recruitment of additional integrins. Indeed, application of force by stretching elastic substrata triggers conversion of unoccupied, low-affinity integrins to a high-affinity state, which induces new binding to the extracellular matrix (12). These reinforcement mechanisms may also mediate changes in downstream signaling. Indeed, the activity of focal adhesion kinase (FAK), a tyrosine kinase that transduces multiple
signals from integrins, requires actinand myosin-dependent tension (13).
Friedland et al. provide evidence that the major fibronectin-binding integrin, α5β1, undergoes a force-dependent conformational transition. This conclusion is based on the ability of integrin α5β1 to be chemically cross-linked to fibronectin. Force is required for conversion of the bond from a non± cross-linkable to
a cross-linkable state, suggesting a change in proximity of key residues.
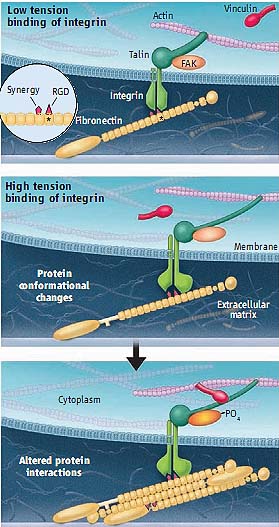
Integrin α5β1 binds to two sites in fibronectin: an Arg-Gly-Asp (RGD)sequence in the 10th type III repeat and a secondary (so-called) synergy site in the 9th repeat (7) (see the figure).
Force mechanics. At points of cell attachment to extracellular
matrix, when tension is low, integrin α5β1 binds to the RGD
sequence in fibronectin. The cytoplasmic domain of integrin α5β1 is associated with talin, but most vinculin binding sites on talin are
inaccessible. Increased tension triggers conformational changes
in the integrin, talin, and fibronectin. These alterations reveal new
vinculin binding sites in talin, enhance the affinity of α5β1 for the synergy site in fibronectin, and reveal new self-association sites in
fibronectin. These changes lead to increased vinculin binding to
talin, tighter binding of the integrin to fibronectin, increased
fibronectin matrix assembly, and activation of FAK. These events
may also promote increased clustering of integrins.
Friedland et al. also show that conversion to the cross-linkable state requires the synergy site and that these events correlate with correlate with phosphorylation (thus, activation) of FAK. The picture that
emerges from these results is that initial low-tension binding of integrin α5β1 to fibronectin involves association of the integrin with the RGD sequence, which under force converts to a higher-strength, more readily cross-linked bond that involves the synergy site. Only this second conformation can activate FAK and transmit downstream signals.
Talin is the focus of the study by del Rio et al. It binds directly to the β1 integrin cytoplasmic domain and forms one of the links to actin. Talin binding is also crucial in the conversion of low-affinity to high-affinity integrins (14) and in the recruitment of vinculin to sites of adhesion (2). Interestingly, the talin rod domain contains multiple vinculin binding sites, but in the intact molecule, most of these sites are buried among bundles of α helices. Indeed, these structural data led to the prediction that unfolding of the talin rod domain under force might expose these cryptic sites to recruit vinculin (15). Because vinculin can also connect to actin, it would reinforce the link between the integrin and the cytoskeleton. Using single-molecule techniques, del Rio et al. show that application of force at the piconewton level results in unfolding of the talin rod domain and binding of vinculin, thus confirming this key prediction.
The advances made by Friedland et al. and del Rio et al. will facilitate answering a number of long-standing questions about integrinmediated mechanotransduction. What is the precise nature of the force-activated conformation of the integrin and how does this conformation promote FAK activation? It' s also not yet clear how recruitment of vinculin to talin mediates downstream signaling, or whether there are other intracellular components recruited to newly exposed binding sites in talin that mediate these functions. Finally, does altered integrin binding to fibronectin directly affect fibronectin matrix assembly, or does this occur through the previously described unfolding of fibronectin domains under force and subsequent self-association (6)? Growing evidence suggests that the entire adhesion apparatus functions as a force-transducing and -sensing machine. These two studies bring us several steps closer to understanding its detailed dynamics.





 Force mechanics. At points of cell attachment to extracellular
matrix, when tension is low, integrin α5β1 binds to the RGD
sequence in fibronectin. The cytoplasmic domain of integrin α5β1 is associated with talin, but most vinculin binding sites on talin are
inaccessible. Increased tension triggers conformational changes
in the integrin, talin, and fibronectin. These alterations reveal new
vinculin binding sites in talin, enhance the affinity of α5β1 for the synergy site in fibronectin, and reveal new self-association sites in
fibronectin. These changes lead to increased vinculin binding to
talin, tighter binding of the integrin to fibronectin, increased
fibronectin matrix assembly, and activation of FAK. These events
may also promote increased clustering of integrins.
Force mechanics. At points of cell attachment to extracellular
matrix, when tension is low, integrin α5β1 binds to the RGD
sequence in fibronectin. The cytoplasmic domain of integrin α5β1 is associated with talin, but most vinculin binding sites on talin are
inaccessible. Increased tension triggers conformational changes
in the integrin, talin, and fibronectin. These alterations reveal new
vinculin binding sites in talin, enhance the affinity of α5β1 for the synergy site in fibronectin, and reveal new self-association sites in
fibronectin. These changes lead to increased vinculin binding to
talin, tighter binding of the integrin to fibronectin, increased
fibronectin matrix assembly, and activation of FAK. These events
may also promote increased clustering of integrins.