Слипчивые клетки поляризуются за счет примыкающего к кости (apical) растяжения мембраны. Эта мембрана, наз. ruffled (рифленный) край, обязана своим видом на поперечном сечении, является каркасом для больших количеств вакуолярных H
-ATPase, которые управляют секрецией кислоты. Вакуольная H
-ATPase экспрессируется в ацидифицированных вакуолях и др. мембранах, транспортирующих кислоту, включая дистальные почечные канальцы, но форма из остеокластов имеет уникальную субъединицу, которая необходима для её экспрессии в ruffled мембране, a3 субъединица (TCIRG). Вакуольная, электрогенная H
-ATPase качает протоны поперек рифленого края, вызывая падение общесредовой pH внутри резорбтивных лакун (Silver, Murrills & Etherington, 1988). Это растворяет костный минерал и позволяет энзимам с кислым оптимумом, таким как cathepsin K, расщеплять коллаген и высвобождать пептиды, которые подвергаются трансцитозу и выбрасываются на дорсо-латеральной поверхности остеокластов. Костный коллаген жестко связан поперечно и пептиды содержат поперечные связи, не деградируемые с помощью proteinases. Количество этих содержащих поперечные связи фрагментов в сыворотке и моче может быть использовано для определения скорости резорбции кости. Электрогенная активность H
-ATPase сбалансирована за счет транспорта анионов. Существует несколько транспортеров анионов в мембранах остеокластов, и недавно было установлено, что недостаточность широко экспрессируемого chloride транспортера, GLCN7 вызывает остеопетроз (Kornak et aL, 2001). CLCN7 является почти определенно chloride-proton antiporter скорее, чем chloride канал, исходя из свойств гомологов из того же самого семейства (Picollo & Pusch, 2005). Это требует, по крайней мере, двух chloride транспортеров для секреции кислоты остеокластами, т.к. chloride-proton antiporter д. функционировать без H
градиента (Diewald et aL, 2002).
Остеопетроз группа родственных болезней с дефектами резорбции кости. Это ведет к накоплению плотного минерализованного хряща с первичным костным матриксом. В своей тяжелой форме болезнь устраняет образование костного мозга с extramedullary гематопоэзом и анемией и вызывает перинатальную слепоту из-за неспособности ремоделирования трактов зрительного нерва. При менее тяжелых формах сохраняется частичная функция остеокластов, совместимая с жизнью, но с ломкими "мраморными костями", склонными к переломам (Li et aL, 2006). Как отмечалось выше основной причиной остеопетроза у человека являются дефекты функции остеокластов скорее, чем их образования, имеется однако множество описаний osteoclast-poor osteopetrosis, но они не были ещё отслежены до специфических онтогенетических генных дефектов. Примерно половина из тяжелых случаев остеопетроза имеет дефекты в a3 субъединице V-ATPase TCIRG1 или в chloride транспортереr CLCN7 (CLC-7). Сегодня единственной эффективной терапией тяжелого остеопетроза является трансплантация костного мозга. Её использование, сопряженное с высокой смертностью из-за ограничения в приживлении, и graft-
versus-host болезни, ограничено случаями, при которых ожидается фатальный исход в отсуствие лечения (infantile malignant osteopetrosis). Pycnodysostosis или болезнь Toulouse-Lautrec, является менее тяжелой sclerotic болезнью. Она вызывает нарушения роста и ломкость костей из-за дефицита активности cathepsin К с плохой деградацией матричных белков. Др. случаи легкого остеопетроза включают некоторые типы недостаточности carbonic anhydrase IIс легким остеопетрозом и дефектами в ацидификации мочи. В остеокластах carbonie-anhydrase-катализируемое превращение CO
2 в carbonic кислоту, увеличивает доступность кислоты и базовых эквивалентов для деградации кости и для chloride-bicarbonate обмена, поддерживает в остеокластах внутриклеточный pH во время секреции кислоты. Редкий синдром с нарушенной резорбцией кости вызывается мутацией стоп кодона в гене
IKKγ . Это нарушает передачу сигналов NF-kB , вызывая anhidrotic ectodermal dysplasia, иммунонедостаточность и остеопетроз (Doffmger et aL, 2001).
V. REGULATION OF BONE RESORPTION BY LOCAL SIGNALS
Тонкий баланс между формированием кости и относительно быстрым процессом резорбции является обязательным условием для поддержания целостности скелета во время взрослой жизни. Как коротко упоминалось в Section I, при hypocalcemia и metabolic acidosis скелет приносится в жертву поддержания кальция и pH в сыворотке даже если это доводит скелетные массы до уровней, вызывающих переломы (Fitz et aL, 1907). Этот факт, который известен более века, часто не принимается во внимание, но он отражает быструю потерю кости при многих типах метаболического ацидоза. PTH составляет исключение, которое подчеркивает правило дифференцировки костных клеток. PTH управляет формированием остеокластов и матричным синтезом остеобластами в качестве генерального ускорителя. Растворение матричного минерала преобладает, но остеокласты не реагируют на PTH, этот механизм обеспечивается посредством остеобластов и их предшественников, реагирующих на растворимый PTH вторичным синтезом RANKL (Huang et aL. 2004), помимо многих др. индуцируемых PTH изменений. Примечательно, в частности, что PTHrP, родоначальный предшественник PTH, интегрируется в и существенен для формирования скелета (Kronenberg, 2006; Guo et aL, 2006). Т.о., растворимый PTH формирует чистый "accelerator", поскольку он использует предсуществующий сайт-специфический механизм, который позволяет контролировать и на организменном уровне. Сходным образом, воспалительный рост, дифференцировка и апоптозный фактор TNFα участвуют в системных заболеваниях, связанных с воспалением, включая ревматоидные артриты и возможно некоторые формы остеопороза и он может появляться в достоверных количествах, как растворимый фактор, хотя почти все TNFs (исключения составляют VEGI/TL1A, который не имеет трансмембранного домена, и TNFα, который не удаляется эффективно) появляются как трансмембранные белки и быстро очищаются и удаляются с помощью растворимых "decoy" рецепторов, включая osteoprotegerin, который соединяется с RANKL и TRAIL, если они высвобождаются с помощью активности proteinase (Bodmer, Schneider & Tschopp, 2002). Этот факт часто забывают, поскольку столь много работ проделано с синтетическими белками, а он лежит в основе изысканного сайт-управляемого образования остеокластов (Fig. 2), которые часто наблюдается на одной стороне трабекулы, тогда как остеобласты добавляют кость к др. , перемещая тем самым трабекулу в пространстве, позволяя ей оставаться ориентированной в пространстве вдоль линий сдавливания в кости.
Растворение hydroxyapatite с помощью HCl поднимает концентрацию Ca2+ приблизительно до 40 мМ в пространстве резорбции остеокластами (Silver el al, 1988). Это активирует сенсор Ca2+ на поверхности остеокластов, вызывая высвобождение Ca2+ из внутриклеточных хранилищ, что сопровождается открытием возможности притока Ca2+ (Zaidi, Moonga & Huang, 20Q4; Moonga et al, 2002; Zaidi et al, 1989). Возрастание цитозольного уровня Сa2+ активирует индуцибельную nitric oxide synthase, которая в свою очередь, генерирует nitric oxide локально, позволяя остеокластам отсоединиться и оттянуться назад (Maclntyre et al., 1991). Возобновление резорбции сопровождается Ca2+-обусловленным синтезом и локальным высвобождением interleukin-6 из ингибированных остеокластов (Adebanjo et al., 1998). Interleukin-6 в свою очередь определяет границы этой ингибирующей обратной связи, позволяя остеокластам возобновить резорбцию. Т.к. физиология контроля восприятия Ca2+ vis-a-vis остеокластами установлена, то мы первоначально не отнесли эту функцию к молекулярной сущности. Вряд ли это parathyroid-cell Ca2+ рецептор. Гомологичное клонирование оказалось неспособным идентифицировать сходную молекулу в остеокластах и остеокласты оказываются нормальными у мышей, лишенных parathyroid Ca2+ рецептора. Более того, calcimimetics не нарушают функции остеокластов, а потребности в восприятии Са2+ в паратироидных клетках и остеокластах количественно различны (Zaidi et al., 2004).
Несмотря на это, мы полагаем, исходя из молекулярных и электрофизиологических доказательств, что существует типа 2 ryanodine рецептор, расположенный на мембране остеокластов, который служит как Ca2+ канал и как Ca2+ сенсор (Moonga et at., 2002; Zaidi et al., 1995). Повышенная резорбция кости у CD38 нокаутных мышей подтверждает это мнение. У этих животных cyclic ADP-ribose (физиологический агонист ryanodine рецепторов) снижена (Sun etal., 2003). CD38 является ADP-ribosyl cyclase, которая катализирует превращение промежуточного метаболита NAD+ в циклическую ADP-ribose. Мы установили, что CD38 воспринимает NAD+, генерируемый как следствие метаболической активности в остеокластах, возникающей в результате их интенсивной подвижности и транспорта кислоты. Путем превращения NAD+ в циклическую ADP-ribose (т.е., путем восприятия метаболического истощения) CD38 по существу связывает метаболическую активность с восприятием Ca2+ (Sun et al, 2002). Интересно, что цитокины, такие как TNFα, которые стимулируют резорбцию кости остеокластами, усиливают активность CD38. Это может позволить избыточность, чтобы защитить скелет от избыточной резорбции (Iqbal el ai, 2006a).
Значительное перекрывание существует при регуляции деградации кости с помощью цитокинов, включая про-резорбтивные эффекты IL-1, -6, -11, -18, TNFα, interferon-γ, также как PTH и oestrogen. Большинство цитокинов и гормонов действуют путем регуляции стромальными клетками продукции RANKL или osteoprotegerin. Др., включая IL-6 и IL-11, обладают общими нижестоящими сигнальными молекулами, такими как gp130. В общем, делеция большинства цитокинов не приводит к фенотипическим отклонениям в скелете. Ни один из большинства цитокинов не является хорошим лекарством, направленным на контроль костной массы. Однако , TNFα обычно повышен при некоторых состояниях потери кости и действует как общий ускоритель потери кости и его прямые эффекты на дифференцировку и активность остеокластов важны. Поэтому anti-TNFα терапия используется для лечения кость-повреждающих болезней, хотя это опасно, т.к. TNFα также важен для иммунной функции (Curtis et al., 2007).
VI. REGULATION OF BONE RESORPTION BY NEURAL MECHANISMS
Помимо гомеостаза кальция посредством PTH, и реакции локальных ростовых факторов на онтогенетические и гормональные стимулы нейроэндокринные сигналы регулируют непосредственно скелетную массу. Сюда входит нейрональные разряды и гипоталямическая регуляция (Zaidi, 2005). Эта нейрогуморальная регуляция является наивысшим уровнем регуляции за пределами межклеточных взаимодействий, используемых в развитии. Гомеостаз PTH/calcium и реакция на растяжение поддерживают микроархитектуру кости. Гипоталямус регулирует как образование, так и резорбцию кости негативно посредством симпатических разрядов (discharges) под центральным leptinergic контролем. Такие симпатические разряды негативно регулируют выход предшественников остеокластов из костномозговых ниш. Существует незначительное перекрывание в этих механизмах контроля высшего уровня и скелетные фенотипические отклонения возникают в результате дефектов любого из нейроэндокринных компонентов. Существует предположение, что непосредственная beta adrenergic реакция регулирует костную массу, но исследования показывают, что это вряд ли достоверно (Reid et ai, 2005; de Vries et al., 2007).
Демонстрация Karsenty с коллегами, что ЦНС регулирует костную массу (Ducy et al, 2000; Takeda et al., 2002) стала центральной для оценки нейральной регуляции скелета. Стимуляция нейронов, экспрессирующих leptin-рецепторы, с помощью центрально применяемого leptin нарушает формирование кости, редуцирует костную массу. Мыши, лишенные leptin или его рецептора, образуют большие костные массы. Эти находки устанавливают центральную leptinergic регуляцию кости, они подтверждены парабиотическими экспериментами. Не выявлено гуморальных эффектов, обеспечиваемых действием leptin на скелет. Однако рецепторы leptin были идентифицированы на костных клетках, это оставляет возможность, что регуляция периферическим лептином может всё же происходить, хотя продуцируемый на периферии leptin не выявлен. Leptin из жировых клеток супрессирует аппетит посредством гипоталямических рецепторов, чтобы регулировать массу тела. Анти-остеогенное действие центрального leptin не зависит от эффектов leptin на аппетит (Elefteriou et al, 2005). Мыши, лишенные нижестоящих leptinergic сигналов, α-melanocyte stimulating hormone или cocaine- и amphetamine-regulated transcript (CART), не имеют костных фенотипических отклонений (Elefteriou et ai, 2005). Более того, тощие leptin-дефицитные lipodystrophic мыши имеют высокую костную массу, фенотип, идентичный таковому тучных ob/ob мышей. Т.о., содержание жировой ткани не влияет на костную массу, по крайней мере, на этих моделях. Более того, ob/ob мыши обладают высокой костной массой несмотря на наличие hypercortisolism и hypogonadism. Эти данные строго подтверждают гипотезу, что эффекты центрального leptin не зависят от половых стероидов и продукции глюкокортикоида.
Дополнительные исследования показали, что центральный leptinergic контроль осуществляется посредством периферических симпатических разрядов (discharges), которые модифицируют активность остеобластов (Takeda et at, 2002). Мыши, лишенные адренэргического рецептора Adrb2 или синтетического энзима dopamine β-hydroxylase? имеют значительную костную массу и у этих мышей отсутствует ингибирующий эффект на образование кости центрально применяемого leptin (Takeda et aL, 2002; Elefteriou et aL, 2005). Сходным образом, β-adrenergic блокада увеличивает костную массу. Интересно, что Ardb2 нокаутные мыши обнаруживают усиленное образование кости и низкую резорбцию кости. Предполагается, т.к. остеокласты являются производными коротко живущих моноцитов и определенно не иннервированы, то эти эффекты leptin на резорбцию кости являются косвенными (Elefteriou et aL, 2005). Это скорее всего осуществляется за счет цитокинов, модулирующих остеокласты, в стромальных компонентах или в самих остеобластах. Т.к. точный механизм неизвестен, интересна связанная находка, что выход гематопоэтических стволовых клеток из костного мозга модулируется симпатическими разрядами, направленными на остеобласты (Katayama et aL, 2006). При потере симпатической активности у мышей, дефицитных по UDP-galactose: ceramide-galactosyltransferase, развитие предшественников остеокластов ослабляется. Т.о., leptinergic нейроны регулируют образование и резорбцию кости вместе с периферическими симпатическими нервами.
Предположено частие циркадного ритма в обороте кости путем адренэргической стимуляции, взаимодействующей с генами часов Per и Cry в остеобластах (Fu et aL, 2005). Мыши, лишенные Per или Cry, или с Per , делетированным только в остеобластах, имеют высокую костную массу и парадоксальное увеличение плотности кости, возникающее после центрального применения leptin. Эти гены обеспечивают антипролиферативное действие и симпатическое воздействие за счет ингибирования G1 cyclin (Fu et aL, 2005). Эти находки могут объяснить суточные вариации в резорбции кости у человека (Qyist et aL, 2002). Пока неясно, однако как центральная нейральная регуляция костной массы обеспечивается исключительно leptinergic и симпатическими механизмами. Anticholinergic и peptidergic нервы также могут участвовать. В самом деле, кости иннервируются Ad- и C-типа нервными волокнами, которые несут некоторые нейротрансмиттеры, включая neuropeptide Y (NPY) и calcitonin gene-related peptide (CGRP). CGRP обладает anabolic и антирезорбтивным действием (Zaidi et aL, 1987), тогда как роль NPYergic нервных окончаний не установлена (Baldock et aL, 2002; Elefteriou et at, 2003). Во всяком случае, потенциал для блокирования симпатических разрядов с помощью избирательного таргетинга кости β-,блокаторами остается. Было бы интересно проверить в клинике будут ли пациенты, получающие β-,блокаторы иметь повышенную костную массу.
Др. нейропептид, который также может экспрессироваться в эндокринных органах и который может регулировать скелетный гомеостаз, это calcitonin. Он вместе с паратироидным гормоном появляется у костистых рыб, чтобы регулировать резорбцию кости. В случае calcitonin, действие завершается активностью остеокластов. Calcitonin связывает определенный G-protein-coupled рецепторы на остеокластах. Он появляется у примитивных организмов, которые имеют предшественник минерализованного скелета, включая асцидии
Stycia clava (Thorndyke and Probert, 1979). Calcitonin у высших позвоночных продуцируется G-клетками щитовидной железы, которые дифференцируются из клеток нервного гребня. Однако до сих пор неясно. является ли calcitonin функциональным антирезорбтивным гормоном у человека. Его делеция у мышей дает высокую скелетную массу скорее, чем остеопению (Zaidi, Moonga & Abe, 2002; Hoff et aL, 2002). Несмотря на это calcitonin эффективно используется как лекарство костных болей у людей. Однако парадоксально у пациентов с imedullary thyroid carcinoma, циркулирующий в крови calcitonin сильно увеличен, но увеличения костной массы не происходит. И у thyroidectomized пациентов с отсутствием практически циркулирующего calcitonin не развивается остеопороз. Т.о., у человека реакция костных болей на calcitonin возможно обусловлена главным образом реакцией центральных нервов, с непосредственным эффектом на оборот кости,
in vivo, возможно являющимся остаточным. Мыши, дефицитные по amylin, родственному с calcitonin пептиду, дают остеопению (Dacquin et aL, 2004; Datta et aL, 1989).
VII. PITUITARY GLYCOPROTEINS AND THE REGULATION OF BONE MASS
Гипофизарная эндокринная ось также стимулирует образование кости посредством оси growth hormone/IGF-1 axis, и регулирует резорбцию кости позитивно и негативно посредством follicle stimulating hormone (FSH) и thyroid stimulating hormone (TSH). В частности гормоны из передней части гипофиза, гормон роста, TSH и FSH, также регулируют костную массу. Др. гормоны из периферических эндокринных органов 1,25-dihydroxyvitamin D3 и тироидный гормон описаны в др. месте (Mundy & Guise, 1999). Гормон роста является anabolic, но осуществляет свои эффекты посредством insulin-like growth factor-1 (IGF-1) из печени. Имеется прекрасная корреляция между костной массой и уровнями IGF-1 в сыворотке. чем между костной массой и гормоном роста в сыворотке (Yakar et aL, 2002). Генетический дефицит IGF-1 вызывает выраженную задержку роста и остеопению (Mohan and Baylink, 2005). Эти скелетные потери необратимы с помощью гормона роста, это подтверждает, что IGF-1 является активным физиологическим агентом. Однако инсулиновые рецепторы не влияют существенно на развитие кости (Irwin et al., 2006), хотя insulin receptor substrate-1 важен для оборота кости (Ogata et al.. 2000), а агонист peroxisome proliferator-activated recepior-Y rosigliiazone снижает образование кости и костной массы (Grey et al., 2007), так что этот вопрос сложен. В противовес непрямому действию гормона роста, устранение гена направляет эффекты TSH и FSH на резорбцию кости (Zaidi et aL, 2006). До сих пор ожидается, что эти гормоны сами регулируют секрецию thyroxine и половых стероидов в эндокринных органах мишенях. В частности, TSH снижает образование, функцию и жизнеспособность остеокластов путем воздействия на G-protein-coupled рецепторы на остеокластах (Abe et al., 2003). То, что этот скелетный эффект является доминантным и выраженным иллюстрируется тем, что вызываемые дефектом TSH рецептором гетерозиготные мыши являются osteopenic несмотря на нормальную функцию щитовидной железы (Zaidi et al., 2006). Сходным образом, FSH стимулирует образование и функцию остеокластов посредством FSH рецепторов на остеокластах, с высокой костной массой у гетерозиготных FSHβ нокаутов. которые имеют нормальную овариальную функцию (Sun et at, 2006).
Как TSH, так и FSH регулируют активность остеокластов ниже RANK, хотя точные механизмы изучены недостаточно (Abe et al., 2003; Sun et al., 2006). Далее экспрессия гипофизарных гликопротеиновых рецепторов в кости, хотя и продемонстрирована четко, может функционировать только в ограниченных условиях и может обнаруживать существенные межвидовые вариации (Blair, Wells & Isales. 2007). Главные механизмы кандидаты включают ингибирование с помощью TSH внутриклеточной киназы Janus N-terminal kinase (JNK) и предупреждение проникновения в ядро с-jun. Напротив, FSH стимулирует фосфорилирование extracellular signal-regulated protein kinase (Erk) и protein kinase В (Akt1) посредством ингибрующего G белка Gi2α. Пептиды противоположным образом затрагивают активацию NF-kB с помощью модулирования фосфорилирования его ингибирующей субъединицы I-кВ. Далее, TSH и FSH оказывают реципрокные эффекты на продукцию TNFα: TSH ингибирует, а FSH стимулирует TNFα Эти действия вносят вклад в анти- и про-osteoclastic действия двух гормонов ( Hase et al., 2006; Iqbal et al., 2006b), и могут иметь ключевое значение.
VIII. CONCLUSIONS
In bone development, local growth and differentiation factors establish the skeletal shape and the characteristics of its cell and tissue components.
Skeletal development follows a sequence that includes conserved elements of early differentiation for segmentation, regulated by hox genes.
Modeling of the skeleton uses a number of gradient sensitive mechanisms and seven transmembrane-pass protein recognition receptors, including PTHrP, Writ, and BMP signals, to regulate where skeletal elements are formed, and where mesenchymal tissue undergoes apopto-sis to form joint spaces.
All new bone is produced and destroyed by new cells differentiating from stem cells. This is a continuous process that occurs throughout life. Thus, the regulation of skeletal turnover, an important biological and medical issue, is logically considered in the context of differentiation.
The growdt factors that control the development of bone also regulate skeletal replacement and use of skeletal mineral for metabolic homeostasis, in some cases by direct adaptation such as the use of PTH for regulation of bone degradation rate using receptors also involved in bone modeling.
While programmed development and local regulation are critical to bone modeling and to regulation of turnover, neural and neuroendocrine regulation of bone integrates bone metabolism with activity of the organism as a whole. These mechanisms reflect, in part, direct signalling by pituitary glycoproteins that are largely known from specialised endocrine regulation but where more primitive direct regulation of skeletal metabolism is retained.
Сайт создан в системе
uCoz Mone Zajdi, Christopher L.-H. Huang and Li Sun
Mone Zajdi, Christopher L.-H. Huang and Li Sun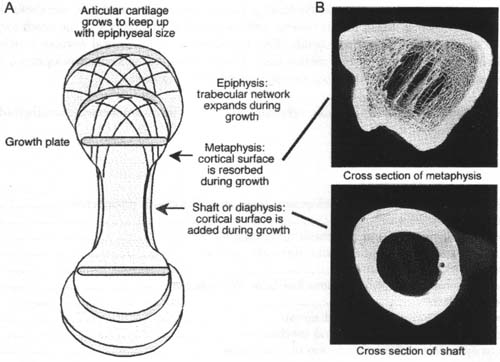 Fig. 1. Development of a long bone in the terrestrial vertebrates. (A) Cartilage from the anlage of the bone is replaced, except at growth plates and articular cartilages, by bone. Bone forms on columns of mineralized cartilage produced during the nansition of cartilage to bone, mediated by differentiation of cells from vascular ingrowth. (B) Trabecular bone reinforces bone in areas subject to complex forces, while in the shafts of long bones it is resorbed. The trabecular bone has a large surface area, and all formation and resorption take place at the bone surfaces. This renders the regions of trabecular bone susceptible to damage when bone mass is lost overall.
Fig. 1. Development of a long bone in the terrestrial vertebrates. (A) Cartilage from the anlage of the bone is replaced, except at growth plates and articular cartilages, by bone. Bone forms on columns of mineralized cartilage produced during the nansition of cartilage to bone, mediated by differentiation of cells from vascular ingrowth. (B) Trabecular bone reinforces bone in areas subject to complex forces, while in the shafts of long bones it is resorbed. The trabecular bone has a large surface area, and all formation and resorption take place at the bone surfaces. This renders the regions of trabecular bone susceptible to damage when bone mass is lost overall. 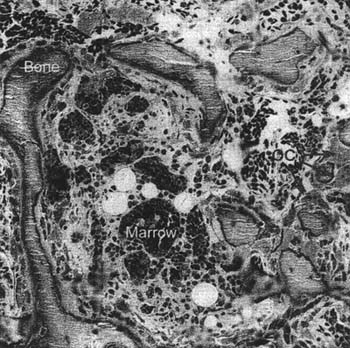 Fig, 2. Bone cell differentiation. Note the distinct compartments of marrow cells producing leukocytes and red blood cells (marrow) as well as bone bordered by osteoblasts (OB) and osteoclasts (ОС). The individual domains are completely separate and highly organised. The field is 0.4 mm square. To minimize arrifacts, a 5 mm cube of live avian trabecular bone was dropped into ice-cold 4% glutaraldehyde and plastic embedded. The tissue was cut without decalcification to 500 nm thickness and stained with Methylene Blue.
Fig, 2. Bone cell differentiation. Note the distinct compartments of marrow cells producing leukocytes and red blood cells (marrow) as well as bone bordered by osteoblasts (OB) and osteoclasts (ОС). The individual domains are completely separate and highly organised. The field is 0.4 mm square. To minimize arrifacts, a 5 mm cube of live avian trabecular bone was dropped into ice-cold 4% glutaraldehyde and plastic embedded. The tissue was cut without decalcification to 500 nm thickness and stained with Methylene Blue. 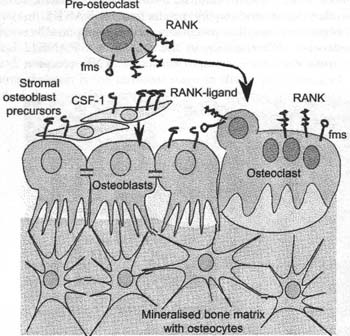 Fig. 3. Mediation of bone cell differentiation by local expression of growdi and differentiation factors. Mesenchymal stem celLs from stroma differentiate to form osteoblasts; this requires multiple stimuli including bone morphogenetic proteins (BMPs) which are not well defined. Osteoblasts are connected in cohorts of cells that secrete matrix togedier by coimexons with CX43 being a central component; lines of cells that are buried in matrix (osteocytes) remain connected to the surface las'er and survive as long as the bone matrix is retained; these cells are believed to be important in stretch-mediated signalling for matrix maintenance. In the case of osteoclast development, however, it is predominately membrane forms of CSF-1, the f'ms ligand, and the tumour necrosis factor (TNFj-famify protein RANKL, which mediate highly localised development of osteoclasts precisely where the inducing growth factors occur.
Fig. 3. Mediation of bone cell differentiation by local expression of growdi and differentiation factors. Mesenchymal stem celLs from stroma differentiate to form osteoblasts; this requires multiple stimuli including bone morphogenetic proteins (BMPs) which are not well defined. Osteoblasts are connected in cohorts of cells that secrete matrix togedier by coimexons with CX43 being a central component; lines of cells that are buried in matrix (osteocytes) remain connected to the surface las'er and survive as long as the bone matrix is retained; these cells are believed to be important in stretch-mediated signalling for matrix maintenance. In the case of osteoclast development, however, it is predominately membrane forms of CSF-1, the f'ms ligand, and the tumour necrosis factor (TNFj-famify protein RANKL, which mediate highly localised development of osteoclasts precisely where the inducing growth factors occur. 
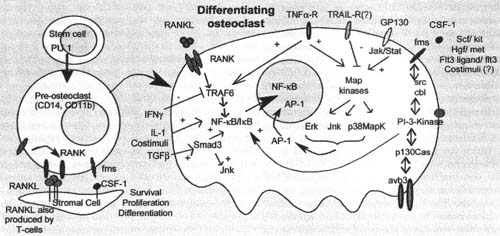 Fig. 4. Selected osteoclast differentiation pathways. In vivo, normal signals for osteoclast differentiation are mostly cell-bound proteins. In vitro, media with selected serum, ligands for Fms and RANK may be sufficient for differentiation, although activation of Fey is also required as a co-stimulus (not diagrammed, see text). Many intermediate steps and cross-relationships of pathways exist in addition to those diagrammed. Some major intracellular proteins are indicated, but many steps in addition to the molecules named are involved; for example, numerous steps, including ubiquitination of IkB and proteasomai degradation, are required for NF-кВ activation.
Fig. 4. Selected osteoclast differentiation pathways. In vivo, normal signals for osteoclast differentiation are mostly cell-bound proteins. In vitro, media with selected serum, ligands for Fms and RANK may be sufficient for differentiation, although activation of Fey is also required as a co-stimulus (not diagrammed, see text). Many intermediate steps and cross-relationships of pathways exist in addition to those diagrammed. Some major intracellular proteins are indicated, but many steps in addition to the molecules named are involved; for example, numerous steps, including ubiquitination of IkB and proteasomai degradation, are required for NF-кВ activation.