Белки, участвующие в архитектуре хроматина, также влияют на обеспечение взаимодействий между хромосомными регионами. Напр., T
2 cytokine локус, SATB1 обеспечивает ассоциации между регионами в цис-положении, чтобы генерировать трехмерную, активную конформацию хроматина [ 29 ]. H19 импринтинг контролирующий регион ассоциирует со множественными геномными локусами, в основном посредством материнского аллеля, который связывает хроматин инсуляторный белок CTCF [ 38 ]. Интересно, что недавние исследования выявили, что связывание CTCF и cohesin, белкового комплекса, известного своей важной ролью в слипчивости сестринских хроматид [ 53 ], перекрывает множественные сайты в геноме человека и мыши [ 54,55 ]. Cohesin и CTC F кооперируют, обеспечивая
[ 56-58]. Можно предположить, что cohesin использует свою способность сводить хромосомные регионы вместе для установления и/или поддержания др. внутрихромосомных и потенциально межхромосомных ассоциаций.
Как геномные регионы 'находят' др. др. и/или ядерные компартменты в сложной ядерной среде, чтобы установить хромосомные ассоциации? В целом перемещения хроматина являются регионально ограниченными в ядре [ 59 ]. Однако это не исключает возможности, что геномные регионы опробуют свою ядерную среду в результате Броуновского движения на относительно коротком расстоянии с последующей стабилизацией предпочтительных ассоциаций. Активные направленные дальнодействующие перемещения хроматина также были описаны. Доставка транскрипционного активатора на массив трансгена приводит к релокализации с ядерной периферии внутрь на расстояние до 5 µm[60]. После индукции транскрипции наблюдаются перемещения более, чем на 2- 3 µm в направлении телец Cajal для U2 snRNA трансгена [ 61 ]. Интересно, что actin [60,61 ] andmyosin[60] участвуют в этих перемещениях хроматина. Сходным образом межхромосомная ассоциация между estrogen-регулируемым
TFF1 и GREB1 генами зависит от actin, nuclear myosin I и dynein light chain-1 (
DLC1) [16], а вмешательство в полимеризацию актина или функцию ядерного myosin I устраняет межхромосомные взаимодействия между связанными с андрогеновым рецептором генами
TMPRSS 2 и ETV1 [52]. Многочисленные исследования наблюдали роль транскрипции ядерного actin и myosinin [62 ], но как actin/ myosin система механистически участвует в перемещении генов и транскрипции пока неясно. Обработка химическими соединениями, которые ингибируют полимеризацию или деполимеризацию, мешают межхромосомным ассоциациям между с ядерными рецепторами связанными генами [ 16,52], а избыточная экспрессия не способного к полимеризации мутантного актина устраняет взаимодействие между тельцами Cajal и U2 массивом [ 61 ], указывая тем самым, что актиновые филаменты могут участвовать в этих перемещениях. Длинные актиновые филаменты сравнимые с теми, что обнаруживаются в цитоплазме, не обнаруживаются в ядрах млекопитающих. Это , однако, не исключает существование относительно коротких, высоко динамичных актиновых филамент, на которые может действовать ядерный миозин, чтобы способствовать направленным перемещениям генов.
Too close for comfort: translocations and trans-splicing
Соприкосновение активных генов может максимализовать результаты транскрипции или делает возможным их ко-регуляцию, но не без риска для клеток. Напр., транслокация prone генетических локусов часто обнаруживается в тесной близи к ядру [ 63 ]. Myc и Igh , частые партнеры по транслокации при лимфоме Burkit t's и плазмацитоме мыши, преимущественно ассоциируют в общей транскрипционной фактории в В лимфоцитах мыши [14 ]. В клетках рака простаты активация транскрипции с андрогенным рецептором связанных генов индуцирует не только их внутрихромосомную [52,64 ] , но межхромосомную [52] ко-локализацию, а также после обработки агентами, которые вызывают разрывы двойной нити ДНК, транслокационные события между этими локусами. Более того, вмешательство в ассоциацию между TMPRSS2 и ETV1 ингибирует транслокацию между двумя локусами [52]. Эти находки в целом указывают на то, что колокализация транскрибируемых генов предоставляет возможность возникновения хромосомных транслокаций.
Было также предположено, что близость активных генов в общих транскрипционных факториях может облегчать trans-splicing [65,66 ], процесс, при котором экзоны от отдельных premRNAs объединяются, чтобы создать химерные РНК. Впервые открытый у трипаносом [67], транс-сплайсинг также существует у млекопитающих и может использовать последовательности от одной и той же хромосомы [ 68,69,70 ], или локализоваться на разных хромосомах [ 71,72]. Для большинства trans-spliced продуктов доказательства функциональной роли отсутствуют. Однако способность транс-сплайсинга дополнять генетические мутации [73 ] была использована для стратегии генной терапии [74,75,76 ], и было продемонстрировано существование важных механизмов для функции генов. Недавнее сообщение описывает чёткую корреляцию между транслокациями и транс-сплайсингом [ 72]. В стромальных клетках человека транс-сплайсин соединяет экзоны от JAZF1 и JJAZ1 генов, продуцируя химерную РНК, которая транслируется в белок с антиапоптической функцией. Удивительно, химерная РНК и белок идентичны тем, что генерируются с помощью транслокаций, обнаруженных в стромальных опухолевых клетках. Единственным возможным объяснением этого является то, что транс-сплайсинг может предрасполагать геномные локусы к хромосомным обменам [72]. Альтернативная возможность состоит в том, что пространственная близость между двумя локусами делает возможным продукцию химерных РНК с помощью транс-сплайсинга в нормальных стромальных клетках, тогда как наложение оказывается зафиксированным посредством транслокаций в некоторых клетках, это позволяет им пролиферировать как раковые клетки. Согласно этому сценарию общий denominator лежащий в основе генерации химерных JA ZF1 - JJAZ1 РНК в нормальных и раковых стромальных клетках, д. находиться в тесной близости в ядерном пространстве, возможно в общей транскрипционной фактории (Figure 1 b,c). Поразительно, что геномная конформация, которая увеличивает риск потенциально важных транслокаций, может эволюционно персистировать. Мы полагаем, что образование трехмерных генных кластеров из транскрибируемых локусов может извлекать эволюционные преимущества, которые перевешивают вред транслокаций.
Conclusions and outlook
Fuelled by the 3C assay [10] and its modifications, our understanding of genome structure and function has remarkably expanded over the past five years. Novel genome-wide proximity ligation assays such a s Hi-C [77] and ChIA-PET [17] now offer the possibility of mapping whole genome conformations. These ‘anchorfree’ assays have the potential to describe connectivity between all loci in the genome, albeit, compared to analyses of chromosomal associations focusing on specific bait loci [13,38,43], this m ay currently come at the expense of a reduced resolution for specific interactions. Nevertheless, aided by the ever-increasing power and rapidly falling cost of high-throughput DNA sequencing, the characterization of the complete repertoire of chromosomal interactions within a cell type now seems an achievable goal. However, caution must be applied when using 3C approaches to study the dynamics of genome organization. Active genes are transcribed in non-synchronous bursts [78,79], and transcription f actory associations between genes in a preferred network vary strongly from cell to cell [43]. This suggests that the transcriptional interactome is inherently plastic and that a ‘single solution’ describing the complete spatial arrangement of the genome in a particular cell type does not exist. Thus, one inevitable caveat of 3 C assays, because they describe the average conformation in a population of cells, is their failure to account for cell-to-cell heterogeneity.
We propose that spatial clustering between co-regul ated genes is a widespread phenomenon. Three-dimensional gene clustering is not only limited to RNAPII tran scription units, but has also been described for genes transcribed by RNA PIII [ 80 ] and RNA PI—in fact, the nucleolus can be regarded as the arche typical example of a specialized transcription factory [81 ]. Other types of specific or preferred interactions are thought to mediate transcriptional repression [20,21 ]. Further more, during immunoglobulin recombination in B cell development [ 19 ], and at the onset of X chromosome inactivation [ 23,24 ], transient interactions between homologous chromosomal regions are involved in establishing polar opposite states of transcriptional activity on homologous alleles, or indeed entire chromosomes. We predict these multiple dynamic chromosomal interactions will together drive higher order chromosome conformations, and tissue-specific chromosome positioning [82 ]. Alteration of gene expression programs during differentiation, development , and nuclear reprogramming [ 83 ] will probably be associated with and may require corresponding changes in the nuclear interactome. A major challenge will be to decipher the relation between these genome conformation changes and the numerous epigenetic alterations of the genome, allowing their integration into a comprehensive picture of the spatial and functional organization of the nucleus .
Сайт создан в системе
uCoz
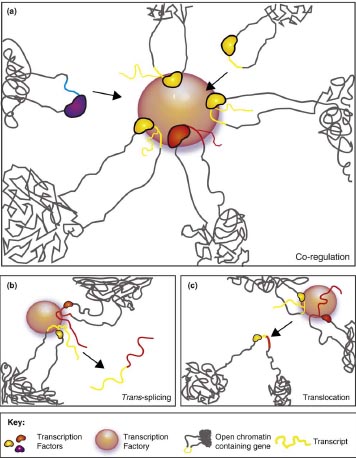 Proximity of active genes in a shared transcription factory. (a ) Co-regulated genes cluster in a specialized transcription factory. Transcription factors (yellow, red, and blue) bind their target Hgenes while probing their nuclear environment. Upon relocation to a transcription factory, potentiated genes initiate transcription (nascent transcripts depicted in yellow and red). Dynamically bound transcription factors may dissociate from their target genes, freeing transcription factors for use by other co-regulated genes in close proximity. Thus, genes in a factory with other co-regulated genes may have a higher probability of re-initiation in that factory through dynamic exchange of transcription factors, stabilizing their presence there. By contrast, genes transcribing in the absence of other network partners (genes regulated by red and blue factors) may be more likely to dissociate from the factory after an initial burst of transcription. Repetition of factor dissociation and binding cycles would result in a transcription site highly enriched in specific binding sites and factors, seemingly specialized to preferentially transcribe a subset of co-regulated genes. (b ) Close proximity between transcripts generated in a transcription factory may allow specific exons to be joined by trans -splicing. (c) Juxtaposition of active genes in a shared transcription factory may also increase the probability of translocations between loci.
Proximity of active genes in a shared transcription factory. (a ) Co-regulated genes cluster in a specialized transcription factory. Transcription factors (yellow, red, and blue) bind their target Hgenes while probing their nuclear environment. Upon relocation to a transcription factory, potentiated genes initiate transcription (nascent transcripts depicted in yellow and red). Dynamically bound transcription factors may dissociate from their target genes, freeing transcription factors for use by other co-regulated genes in close proximity. Thus, genes in a factory with other co-regulated genes may have a higher probability of re-initiation in that factory through dynamic exchange of transcription factors, stabilizing their presence there. By contrast, genes transcribing in the absence of other network partners (genes regulated by red and blue factors) may be more likely to dissociate from the factory after an initial burst of transcription. Repetition of factor dissociation and binding cycles would result in a transcription site highly enriched in specific binding sites and factors, seemingly specialized to preferentially transcribe a subset of co-regulated genes. (b ) Close proximity between transcripts generated in a transcription factory may allow specific exons to be joined by trans -splicing. (c) Juxtaposition of active genes in a shared transcription factory may also increase the probability of translocations between loci.