замалчивается в лимфоидном клоне. Однако пока ген действительно замалчивается эпигенетически из-за недоступности места старта транскрипции и метилирования ДНК промотора в Т клетках, промотор и интронные регуляторные элементы поддерживаются в частично активной конформации хроматина в B-клетках. Это продемонстрировано благодаря непрерывной экспрессии в B-клетках
.
, однако, не экспрессируется в B-клетках благодаря активной супрессии промотора с помощью Pax5, который необходим для поддержания качественных особенностей B-клеток (Tagoh et al., 2006). Поэтому ремоделирование хроматина является динамическим процессом и могут существовать промежуточные состояния с ограниченной способностью клеток к переходу между типами.
Становление одной онтогенетической судьбы обычно препятствует приобретению альтернативной судьбы. Это важно для поддержания структурной целостности в данном пространственном домене. Это достигается становлением множественных межклеточных и внутриклеточных схем регуляторных петель обратной связи, которые активно поддерживают данное состояние и репрессируют альтернативные возможности. Создание помехоустойчивости также защищает от сбоев. Регуляторные sub-circuits, которые специфицируют различные части тела, были отобраны в ходе эволюции, чтобы предоставить максимум страховки от возможности онтогенетических провалов (Cripps and Olson, 2002; Hinman et al., 2003; Pimanda et al., 2007b). Более того, элементы subcircuit любой онтогенетической системы могут отличаться возрастом и источником в эволюции, отобранные и собранные по кусочкам сучше всего подходят к обстоятельствам (Davidson and Erwin, 2006; Tokuoka et al., 2004). Помехоустойчивость достигается также степенью встроенной избыточности. CRMs, которые составляют узлы сети, часто обладают множественными сайтами связывания транскрипционных факторов и используют множественные родственные факторы для управления генной экспрессией.
Network reconstruction
Специфичные для типов клеток сети транскрипционных факторов детерминируют клеточные реакции на внешние сигналы. Современные стратегии разработки технической документации таких сетей распадаются на две категории. Подход восходящего анализа (bottom-up approach) использует ткане-специфические CRMs из ключевых регуляторов данной ткани в качестве построения блок за блоком регуляторных взаимодействий, которые оперируют в этой ткани. Подход нисходящего анализа пытается реконструировать сети из профилей экспрессии по всему геному для определенной ткани и предсказуемых взаимодействий между этими экспрессирующимися генами.
Bottom-up подход наиболее подходит для haematopoietic stem cell (HSC) сети, идентифицированной с использованием Scl/Tal1 гена в качестве стартовой точки. Scl/Tal1 необходим для спецификации развития крови из мезодермальных предшественников. Разрушение этого гена во время развития приводит к отсутствию крови и гибели эмбрионов. Энхансер в 600 п.н. стоящий ниже точки старта транскрипции на +19 т.п.н. оказался достаточным для наделения экспрессией репортерного гена HSCs и эндотелия трансгенных эмбрионов (Gottgens et al., 2002; Rainis et al., 2005; Sanchez et al., 1999; Sanchez et al., 2001; Silberstein et al. , 2005; Sinclair et al., 1999). In-vivo активность, как было установлено, зависит от двух законсервированных Ets-связывающих сайтов и одного законсервированного GATA-связывающего сайта, которые связывают Fli1, Elf1, Erg и Gata2 в предшественниках крови. Энхансер Scl+19 оказался первым in vivo установленным HSC цис-регуляторным модулем любого гена, необходимого для спецификации HSCs. Используя Ets/Ets/Gata конфигурацию Scl+19 энхансера в качестве матрицы, последующий компьютерный скрининг выявил (Donaldson et al., 2005a) 67 кластеров, которые были законсервированы в геномах человека, мыши, собаки с тем же самым пространственным расположением и ориентацией, что и Scl+19. Три из этих с помощью компьютера идентифицированных кластеров находились в Fli1, Hhex и Smad6 генных локусах и обнаруживали HSC активность, сходную родительским Scl+19 энхансером (Donaldson et al. , 2005b, Pimanda et al., 2007a). С помощью биоинформатики, ChIPseq и анализа трансгенов, были добавлены дополнителные регуляторы гематопоэза к выявляемой сети регуляторных генов (Chan et al., 2007; Chapman et al., 2003; Landry et al., 2008; Okuno et al., 2005; Wilson et al., 2009).
Следовательно, воспроизведение сетей с использованием bottom-up подхода осуществляется в несколько этапов. Наиболее важным из которых является идентификация ткане-специфических CRMs генов, необходимых для спецификации этой ткани. За этим следует идентификация мотивов, связывающих транскрипционные факторы внутри этих моделей, которые вожны благодаря их активности и используются в технике, базирующейся на ChIP (Chromatin Immuno Precipitation) , чтобы идентифицировать транскрипционные факторы, которые экспрессируются в этой ткани и соединяются с этими модулями. С этой точки зрения можно разработать или онтогенетическую иерархию путем идентификации ткане-специфических регуляторных модулей транскрипционных факторов, которые являются дайверами родительского CRM или используются в качестве биоинформационного инструмента для предсказания др. мишеней CRMs, которые реагируют параллельно с родительским CRM. Когда подготовлена онтогенетическая иерархия, чрезвычайно важно знание времени осуществления экспрессии различных транскрипционных факторов во время развития в определенной ткани (Patterson et al., 2007; Patterson et al., 2005; Swiers et al., 2006). ChIP-chip анализ соотв. клеток также может быть бесценным в идентификации потенциальных ткане-специфических CRMs в регуляторных генах, которые затем устанавливаются, как настоящие нацеленные на HSCs с использованием трансгенных подходов.
Подход top-down, который был использован успешно для выявления регуляторных сетей у бактерий и дрожжей, недавно был приспособлен к разработке reverse engineer сетей в B лимфоцитах и макрофагах (Basso et al., 2005; Gilchrist et al., 2006). В широком смысле, эти подходы базируются на данных по экспрессии по всему геному в клетках после различных стимулов или экспериментальных условий, таких как точки старта и используемые алгоритмы, чтобы получить информацию о межгенной ко-регуляции. Эти методы имеют целью генерацию графических репрезентаций клеточных сетей, где узлы представляют гены, а соединения (edges) между ними представляют взаимодействия или между кодируемыми белками или между кодируемыми белками и генами (Hartemink et al. , 2001). Подходы top down были по-разному успешны у простых организмов и распадались на две категории: методы оптимизации, которые усиливают функцию количественной оценки относительно моделей альтернативных сетей (Gat-Viks and Shamir, 2003; Gat-Viks et al., 2003); метод регрессии, который сравнивает данные с моделями a priori (Gardner et al., 2003); интегративные биоинформационные подходы, которые комбинируют данные от ряда независимых экспериментальных открытий (Ideker et al., 2001); и статистические методы, которые базируются на измерениях варианс корреляций генной экспрессии парным образом (Butte and Kohane, 2000).
Недавний успех в реконструкции регуляторных сетей у млекопитающих с использованием top-down подхода коснулся B-клеток человека. Используя новый метод, наз. ARACNe (algorithm for the reconstruction of accurate cellular networks), авт. показали, что относительно небольшое число сильно связанных генов взаимодействуют с большинством др. генов в клетке, или непосредственно или иерархически посредством сильно связанных sub-hubs (Margolin et al., 2006). Прото-онкоген MYC был идентифицирован как один из самых крупных ступиц (hubs) в B-клеточной сети (Basso et al., 2005). Подход top-down был также использован, чтобы показать, что activating transcription factor 3 (ATF3) негативно регулирует программу транскрипции Toll-like рецептора в макрофагах (Gilchrist et al., 2006). Авт. проанализировали волны генной транскрипции после стимуляции макрофагов агонистом Toll-like receptor 4 и образование кластеров генов, базируясь на их временных паттернах экспрессии. Они предположили, что кластеры генов, которые включают ATF3 (cluster 6) , чья мРНК достигает пика в течение 1 ч, ко-регулируются и обладают общими CRMs, которые ответственны за один или более членов этого кластера. Сайты связывания ATF, как было установлено, представлены избыточно в этих модулях. Используя Cytoscape, анализ сети и инструмент визуализации, чтобы предсказывать межбелковые взаимодействия, было предположено, что ATF3 действует сочетано с NF-kB и AP1. Путем сканирования последовательностей ДНК для ATF3/ NFkB и AP1 сайтов связывания вблизи к точкам старта транскрипции, IL6 и IL12b были идентифицированы как гены мишени, которые замалчиваются после связывания ATF3.
Существует приблизительно 200 генов транскрипционных факторов, экспрессирующихся в нейральных стволовых клетках и возможно сходные количества в HSCs (Akashi et al., 2003; Kummerfeld and Teichmann, 2006). С помощью вычислений предсказать и экспериментально картировать каждое взаимодействие транскрипционного фактора с ДНК - колоссальная задача. Компьютерные предсказания ограничены тем фактом, что специфичность связывания ДНК лишь только часть функции транскрипционных факторов, которая достаточно охарактеризована, чтобы предсказывать последовательности, которые они могут и не могут связывать, хотя продолжаются попытки исправить это (Berger et al., 2006). Большой набор данных, который будет сгенерирован может быть отфильтрован при сосредоточении внимания на филогенетически законсервированных сайтах связывания, хотя филогенетический footprinting не может идентифицировать сайты, которые функционально не законсервированы (Odom et al., 2007). Наборы данных в дальнейшем могут быть отфильтрованы благодаря включению крупно масштабной верификации доступности хроматина и событий связывания транскрипционных факторов, используя ChIP- chip (Boyer et al., 2005) или ChIP-sequencing технологии (Marson et al., 2008). Основным недостатком, однако, является количество клеток, которое необходимо для осуществления этих экспериментов на геномной шкале, имея в виду, что на ст E11.5 мышиный AGM (Aorta- Gonad- Mesonephros), как подсчитано, обладает приблизительно одной HSC на эмбрион. Другое ограничение в точной идентификации ткане-специфических стволовых клеток. Чувствительность современного протокола проточной цитометрии, используемой для идентификации HSCs приблизительно 50% (Weksberg et al., 2008) для мыши и 1% для человека (Majeti et al., 2007).
Regulatory networks in haematopoiesis
Чтобы определить регуляторное состояние HSC, необходимо сначала установить клетку или клетки из которых они возникают и сайт или места из которых они происходят, а не только располагаются. То, что клетки предшественники, являются производными мезодермы и обнаруживают тесную ассоциацию с эндотелиальным клоном установлено и детально описано в данном приложении (Fig. 4). То, что AGM является местом генерации HSC беспорно и недавно продемонстрировано, что происходит долговременное заселение вновь HSCs в AGM, возникающем из VE-cadherin
+ CD45
+ предшественников (Taoudi et al., 2008). Если свойства HSCs униформны, то можно предположить, что где бы не были или откуда бы не происходили предшественники, состояние хроматина и транскрипционных факторов, которые экспрессируются (с поправкой на фактор избыточности) в этих предшественниках одинаковы. Однако молекулярный и фенотипический анализ четко показывает прирожденные различия между HSC, выделенные от мышиных эмбрионов, молоди, взрослых и старых мышей (Dykstra et al., 2007; Kim et al., 2006; Sieburg et al., 2006; Yilmaz et al., 2006). Ntv не менее, несмотря на эти различия, все эти популяции HSC обладают общими отличительными признаками долговременного самообновления и способности к мультиклональной дифференцировке. Следовательно, вполне возможно, что многие ключевые аспекты
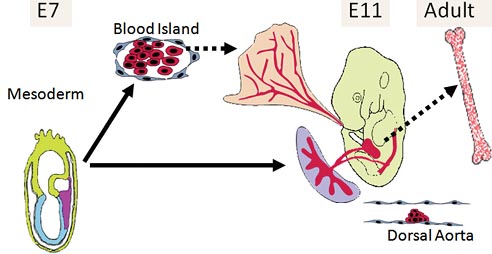 Fig. 4. The ontogeny of blood development in a mouse model. Mesodermal cells from the posterior primitive streak (purple) of day 7 embryos establish extra-embryonic yolk sac endothelial plexus and blood. These cells circulate in the embryo after the establishment of cardiac contractions at day 8. Long-term repopulating blood stem cells originate from and around the dorsal aorta in the AGM region around day 10. These stem cells number is amplified in the fetal liver and placenta and populate the bone marrow around the time of birth. The bone marrow continues as the major haematopoeitic organ in adult life.
Fig. 4. The ontogeny of blood development in a mouse model. Mesodermal cells from the posterior primitive streak (purple) of day 7 embryos establish extra-embryonic yolk sac endothelial plexus and blood. These cells circulate in the embryo after the establishment of cardiac contractions at day 8. Long-term repopulating blood stem cells originate from and around the dorsal aorta in the AGM region around day 10. These stem cells number is amplified in the fetal liver and placenta and populate the bone marrow around the time of birth. The bone marrow continues as the major haematopoeitic organ in adult life.
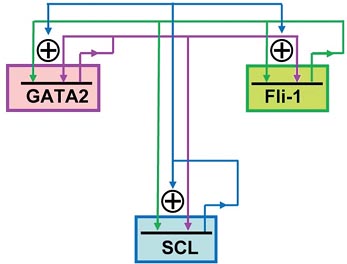 Fig. 5. A fully connected triad of HSC transcription factors. Scl, Fli1 and Gata2 regulate each other by collectively binding the Scl+19, Fli1+12 and Gata2-3, haematopoietic stem cell enhancers. This triad is probably activated by Bmp4 signalling and the expression of these essential haematopoietic factors in HSCs can be maintained by each other in the absence of the activator.
Fig. 5. A fully connected triad of HSC transcription factors. Scl, Fli1 and Gata2 regulate each other by collectively binding the Scl+19, Fli1+12 and Gata2-3, haematopoietic stem cell enhancers. This triad is probably activated by Bmp4 signalling and the expression of these essential haematopoietic factors in HSCs can be maintained by each other in the absence of the activator.
архитектуры GRN законсервированы среди этих разных HSC популяций. Мы сообщали. что Gata2, Fli1 и Scl транскрипционные факторы, каждый из которых экспрессируется в HSCs, перекрестно регулируют др. др. в AGM путем связывания CRMs, которые активны в AGM и в LTHSCs (Pimanda et al., 2007b) (Fig. 5). Эти транскрипционные факторы необходимы для нормального гематопоэза и и располагаются иерархически ниже Bmp4, морфогена, который необходим для спецификации мезодермы и спецификации гематопоэза внутри мезодермы (Maeno et al., 1996). Эта триада транскрипционных факторов является компонентом большой сети в HSCs, которую мы сконструировали, используя bottom up подход. Принципиальная схема между Gata2, Fli1 и Scl и их CRMs соответствует мотиву сети из трех генов, названному полностью соединенной триадой (Pimanda et al., 2007b). Эта трехсторонняя (3-way) позитивная подпитываемая непосредственно петля, которая обнаруживает тенденцию быть закрытой в ON состоянии, редко в экспериментально установленных GRNs бактерий и дрожжей. Важным признаком полностью соединенной триады является то, что вследствие её активации, члены будут увековечивать экспрессию др. др. Следовательно, активирующий сигнал д. быть преходящим, но достаточным для инициации ключевой программы транскрипции, которая буде поддерживаться за счет непрерывного динамического взаимодействия (see preceding section on memory). Это имеет важное значение для клеток, таких как HSCs, которые возникают в одном месте (AGM) и затем путешествуют в др. (печень плода) для дальнейшей амплификации прежде, чем окончательно расположиться в костном мозге.
Триада Gata2/Fli1/Scl также занимательна, поскольку она предоставляет средство для достижения цели, чтобы отслеживать события, которые лежат в основе развития (Fig. 6). Развитие HSCs в AGM регулируется с помощью Notch-1 оно нарушено у Notch-1 дефицитных эмбрионов мыши (Kumano et al., 2003). Notch-1 и Gata2 ко-экспрессируются в гемогенном эпителии в вентральной части аорты на ст. E10.5 и Notch1 связывает промотор Gata2 и действует как вышестоящий регулятор экспрессии Gata2 (Robert-Moreno et al., 2005). Notch 1 сам стоит ниже пути передачи сигналов hedgehog и Vegf (Gering and Patient, 2005; Lawson et al., 2002). Однако, поскольку активация регуляторных элементов зависит от и взаимодействий между Gata2 и Fli1 (Scl скорее всего выполняет количественную роль как только комплекс Gata2/Fli1 будет связан), активация только Gata2 вряд ли достаточна, чтобы инициировать трааду. Передача сигналов Bmp4 необходима для формирования дорсо-латеральной пластинки мезодермы, которая дает дорсальную часть аорты и последующее развитие крови. Разрушение Bmp4 ведет к потере экспрессии
Fli1 и Scl (Walmsley et al., 2002). Промотор Gata2 реагирует на Bmp4, который также инициирует экспрессию
Fli1 (Oren et al., 2005). Активация Gata2 и Fli1 может оказаться достаточной, чтобы запустить схему, которая д. поддерживать экспрессию др. др. и
Scl. Столь же важно как и поддержание активности триады в HSCs является устранение триады, чтобы уменьшить концентрации этих факторов, когда клетки дифференцируются
.
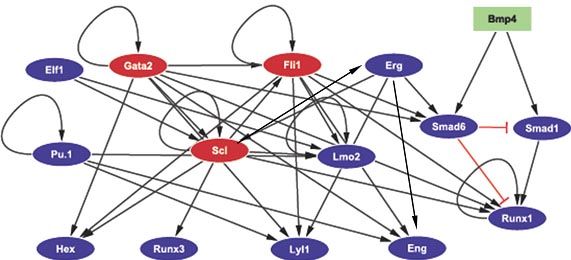 Fig. 6. A bottom-up reconstruction of the haematopoietic stem cell regulatory network. The connections between each of these genes have been experimentally verified.
specific lineages. Interestingly, Gata1 has been shown to disrupt Gata2-positive auto-regulation by binding the Gata2-3 enhancer as cells differentiate down the erythroid-megakaryocytic lineages (Grass et al., 2003). Given the critical and interaction between Gata2 and Fli1, shutting down Gata2 may be sufficient to abrogate the triad and one would predict that maintenance of Scl expression in maturing erythroid cells would require an alternative erythroid enhancer. Interestingly, such an enhancer has indeed been identified and shown to depend on Gata1 and Scl as upstream inputs (Ogilvy et al., 2007).
Fig. 6. A bottom-up reconstruction of the haematopoietic stem cell regulatory network. The connections between each of these genes have been experimentally verified.
specific lineages. Interestingly, Gata1 has been shown to disrupt Gata2-positive auto-regulation by binding the Gata2-3 enhancer as cells differentiate down the erythroid-megakaryocytic lineages (Grass et al., 2003). Given the critical and interaction between Gata2 and Fli1, shutting down Gata2 may be sufficient to abrogate the triad and one would predict that maintenance of Scl expression in maturing erythroid cells would require an alternative erythroid enhancer. Interestingly, such an enhancer has indeed been identified and shown to depend on Gata1 and Scl as upstream inputs (Ogilvy et al., 2007).
Реконструкция основной транскрипционной регуляторной схемы не закончена без включения регуляторных РНК. miRNAs, , по-видимому, регулируют экспрессию существенной пропорции от всех генов в различных типах клеток млекопитающих, включая и гематопоэтические клетки (Garzon and Croce, 2008; Lewis et al., 2005; Stefani and Slack, 2008). Экспрессия miRNAs неслучайна. Эти гены сами по себе участвуют в клеточной дифференцировке и являются объектом ткане-специфической регуляции (Dore et al., 2008). Непоследовательные feed-forward петли, где X активирует Z, но также и репрессирует Z путем активации репрессора Y являются классическим сетевым мотивом, используются для генерации пульсо-образной динамики (Alon, 2007b, Basu et al., 2004). Регуляторные гены miRNA идеально подходят для действия в качестве Y типа репрессоров. В самом деле, путем корреляции по всему геному три-метилирования четвертого остатка лизина Histone H3 субъединиц (H3K4M3 хроматиновая метка) с известными точками старта транскрипции первичных и зрелых miRNA транскриптов и ESTs (а не промоторов белок-кодирующх генов) были сконструированы директории мест старта транскрипции miRNA генов, которые экспрессируются в ES клетках и было продемонстрировано перекрывание расположения Oct4, Sox2, Nanog и Tcf3 на промоторах miRNA (Marson et al., 2008). Т.к. расположение на промоторе Oct4, Sox2, Nanog и экспрессия белок кодирующих генов в ES клетках уже картировано и поскольку экспериментально установлены мишени определенных miRNAs, то стало возможным увязать модуляцию miRNA с сетью регуляторных генов в ES клетках.
Regulatory networks and haematopoietic disorders
Диагностика и классификация гематопоэтических озлокачествлений в основном базируются на морфологии и иммунофенотипе. Хотя определение профилей экспрессии генов гематопоэтических озлокачествлений, базирующееся на молекулярных сигнатурах, оказалось успешным (Golub et al., 1999), вряд ли оно приведет к рутинной диагностике и парадигмам терапии, несмотря на меньшую токсичность и лучшие альтернативы лечения. Если целенаправленная терапия является целью, то профили дифференциальной генной экспрессии сами по себе не очень информативны. Если профили экспрессии могут быть организованы в иерархические interactome, которые потенциально направляют разработку целенаправленной терапии, то успехи профилирования экспрессии станут более осязаемыми. Сложный анализ тканевых регуляторных сетей предоставляет каркас для понимания пертурбаций, которые ведут к различным болезненным фенотипам. Он также наделяет смыслом большинство важных регуляторных генов. Напр., MYC является одной из наиболее соединяемой ступиц (hubs) в B-клеточном interactome (Li et al., 2003) и является ключом онкогенных повреждений при Burkitts лимфоме и потенциальной мишенью для лечения рака (Soucek et al., 2008). Очевидно, если повторяющиеся цитогенетические повреждения могут быть идентифицированы в опухолях или с помощью конвенционного кариотипирования или с помощью сравнительной геномной гибридизации также как и вовлекаемые гены, которые или сильно связаны или являются критическими медиаторами важной ступицы (hub) внутри нормального интерактома, то они и будут мишенями для терапии. Профили экспрессии были также использованы для характеристики молекулярных сигнатур, возникающих после специфических фармакологических вмешательств (Lamb et al., 2006). Опять же было бы намного информативнее охарактеризовать сети, которые нарушаются в ответ на специфические фармакологические воздействия, чтобы они могут привести к терапии. С помощью профилирования экспрессии опухолей и идентификации ключевых сетей, которые оказываются в беспорядке, могут планироваться специфические вмешательства, базирующиеся на прототипах.
Анализ сети гематопоэтических озлокачествлений может быть хорош только как оригинальный образец (template). Наиболее информативным образцом кровяных клеток, кстати, является В-клеточный интерактом, сконструированный с помощью профилей суммарной генной экспрессии на нескольких стадиях В-клеточного развития с известными межбелковыми и белок-ДНК взаимодействиями и известными модуляторами этих взаимодействий скомпилированными из разных баз данных (Mani et al., 2008). Одним из недостатков этой методологии является необходимость в больших background популяциях, важных для сравнения. Поскольку зависимость показателей подобно полному количеству информации нуждается в определенном размере выборки,чтобы добиться достоверности, то это делает затруднительными ситуации, где размеры выборки лимитированы (Mani et al., 2008).
Challenges and future directions
By their very nature, HSCs are suspended in transit towards another more differentiated state. HSC purity and adequate cell numbers for in vitro analyses remain major issues for network reconstruction. In vitro or in vivo culture and expansion of HSCs without altering their phenotype would be necessary unless fundamental technical advances in methodology permit gene expression and ChIP on very small numbers of cells. One possible substitute is a robust ES cell differentiation system in which LT-HSCs can be effectively identified.
To reconstruct networks, effective methods to identify protein-DNA interactions are required. The reliance on specific antibodies for ChIP is a hurdle. Methods to express tagged transcription factors at appropriate concentrations in ES cells to reconstruct the ES cell interactome (Kim et al., 2008; Wang et al. , 2006) have been successful and could be adapted to HSCs derived from ES cells if the necessary protocols are developed. A limitation that is likely to be overcome in the near future is the lack of a comprehensive database of binding specificities of all the 2,500 or so known transcription factors. Protein binding microarray technology is rapidly increasing the number of known binding specificities (Berger et al., 2008; Berger et al., 2006). High throughput sequencing is also likely to have a major impact on our ability to document genome-wide transcription factor binding events using ChIP-seq technology. Transgenic evaluation of CRMs is currently regarded as necessary to determine potential activity in LT-HSCs. This is expensive, labour intensive and time consuming. The evaluation of CRMs in ES cell differentiation assays could be an acceptable alternative. As a clearer map of cell signalling cascades that drive developmental processes evolves, a future challenge will be to integrate haematopoietic transcriptional networks with these cascades that activate the regulatory programme.
Сайт создан в системе
uCoz
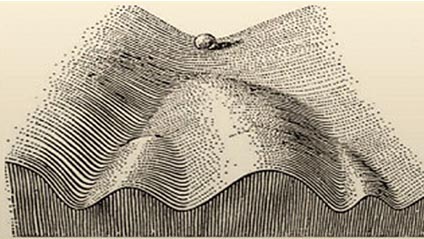 Fig. 1. Conrad Waddington's epigenetic landscape. The ball represents a cell and the bifurcating system of valleys represents trajectories of cell state. This diagram by C.H. Waddington neatly encapsulates the developmental pathways and progressive divergence of cells as they differentiate in the embryo. Reproduced from Waddington, CH © (1957) George Allen and Unwin (London).
Fig. 1. Conrad Waddington's epigenetic landscape. The ball represents a cell and the bifurcating system of valleys represents trajectories of cell state. This diagram by C.H. Waddington neatly encapsulates the developmental pathways and progressive divergence of cells as they differentiate in the embryo. Reproduced from Waddington, CH © (1957) George Allen and Unwin (London). 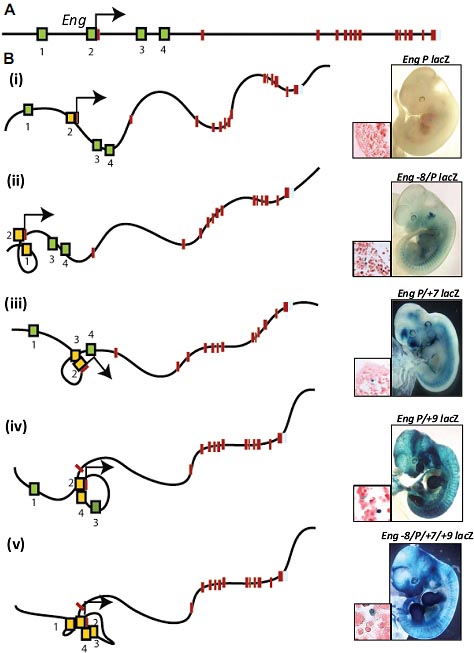 Fig. 2. Distinct enhancers are used to regulate the expression of the same gene in different tissues. (A) The endoglin gene drawn to scale with the promoter (2) and -8kb (1), +7kb (3) and +9kb (4) enhancers marked in green and exons in brown. (B) Transgenic mice generated with Endoglin promoter constructs ( Eng P lacZ) show little endothelial and no blood activity. Transgenic mice generated with the -8 enhancer and endoglin promoter ( Eng -8/P lacZ) shows robust endothelial but no blood activity. Various combinations of the Eng P and Eng-8/P constructs together with the Eng +7 and Eng
+9 enhancers target blood and endothelium.
Fig. 2. Distinct enhancers are used to regulate the expression of the same gene in different tissues. (A) The endoglin gene drawn to scale with the promoter (2) and -8kb (1), +7kb (3) and +9kb (4) enhancers marked in green and exons in brown. (B) Transgenic mice generated with Endoglin promoter constructs ( Eng P lacZ) show little endothelial and no blood activity. Transgenic mice generated with the -8 enhancer and endoglin promoter ( Eng -8/P lacZ) shows robust endothelial but no blood activity. Various combinations of the Eng P and Eng-8/P constructs together with the Eng +7 and Eng
+9 enhancers target blood and endothelium. 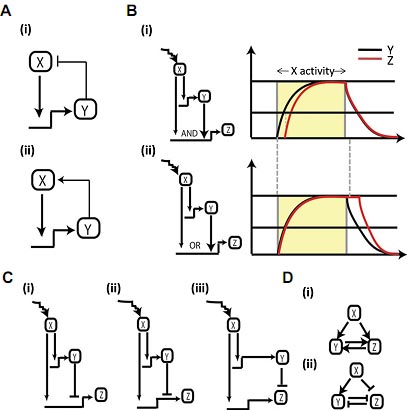 Fig. 3. Different network motifs can be used to regulate the expression kinetics of developmentally important genes. (A) (i) Simple negative regulation; the activation of X, up-regulates Y, which down regulates the activator (ii) Simple positive regulation; the activation of X, up-regulates Y, which further increases X. (B) Coherent feed-forward loops with different logic gates determine rates of target gene expression (i) shows an AND gate with delayed onset and rapid loss (ii) shows an OR gate with rapid onset and delayed loss of target gene expression. (C) Incoherent feed-forward loops, where the activator X up-regulates Z expression but also inhibits Z by up-regulating Y. These incoherent feed-forward loops act at different levels (i) transcription (ii) post-transcription (iii) post- translational. (D) Double positive and negative motifs. (i) Y and Z can continue to up-regulate each other after the activator X has been extinguished. (ii) The inhibition of Z can be initiated by the activation of X and maintained by Y.
Fig. 3. Different network motifs can be used to regulate the expression kinetics of developmentally important genes. (A) (i) Simple negative regulation; the activation of X, up-regulates Y, which down regulates the activator (ii) Simple positive regulation; the activation of X, up-regulates Y, which further increases X. (B) Coherent feed-forward loops with different logic gates determine rates of target gene expression (i) shows an AND gate with delayed onset and rapid loss (ii) shows an OR gate with rapid onset and delayed loss of target gene expression. (C) Incoherent feed-forward loops, where the activator X up-regulates Z expression but also inhibits Z by up-regulating Y. These incoherent feed-forward loops act at different levels (i) transcription (ii) post-transcription (iii) post- translational. (D) Double positive and negative motifs. (i) Y and Z can continue to up-regulate each other after the activator X has been extinguished. (ii) The inhibition of Z can be initiated by the activation of X and maintained by Y.