Специфическая уязвимость определенных зубов связана с их анатомическим положением или на дистальном конце первичной зубной пластинки (primary dental lamina (second premolars)) или в областях слияния лицевых отростков (верхнечелюстные боковые резцы и нижнечелюстные центральные резцы) (Montagu, '45; Svinhufvud et al., '88) и аномалии резцов также наблюдаются как микроформы оральных расщеплений (rev. Ranta, '86). С др. стороны, зубы наиболее часто затрагиваемые, являются последними развивающимися в своем соотв. классе, указывая тем самым, что агенез отражает количественные дефекты во время развития. В терминах теории морфогенетического поля Butler's (1939), агенез может рассматриваться как критическое уменьшение морфогенетического потенциала на периферии поля, вследствие или снижения силы поля в целом или изменения крутизны градиента поля. Аналогично, в терминах клональной теории (Osborn, '78), агенез может быть интерпретирован как следствие снижения онтогенетического потенциала инициальных клонов или изменений в использовании этого потенциала или регулируемых его активации или ингибирования. Уменьшение количества зубов во время эволюции обычно также затрагивает зубы, которые инициируются последними (Miles и Grigson, '90; Weiss et al., '98), указывая тем самым, что механизмы, отвечающие за эволюционные изменения могут также вносить вклад в агенез зубов. В самом деле, некоторые авт. рассматривают зубы, которые подвергаются агенезу, как рудиментарные органы или связывают агенез зубовs с предстоящими эволюционными изменениями (Clayton, '56; Vastardis, 2000). Понимание агенеза зубов как результата количественных дефектов позволяет предполагать участие количественной мультифакторной этиологии, но, как будет показано ниже, существует также врожденная составляющая для молекулярного патогенеза моногенного наследования.
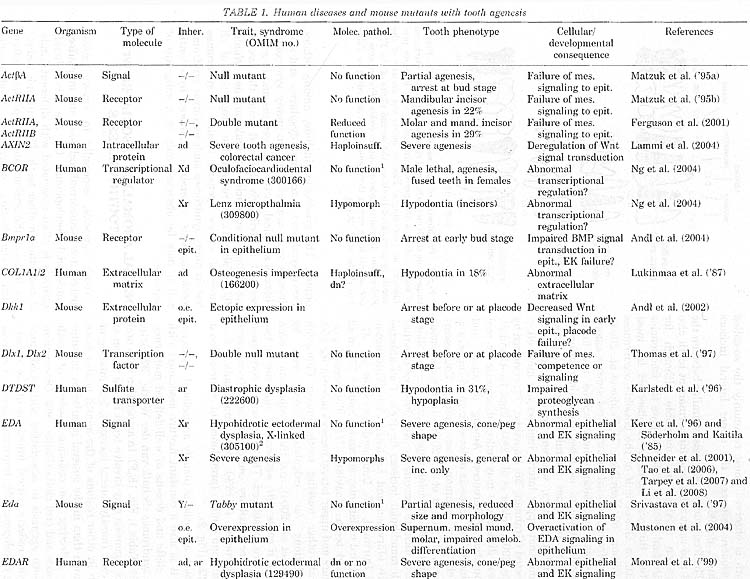
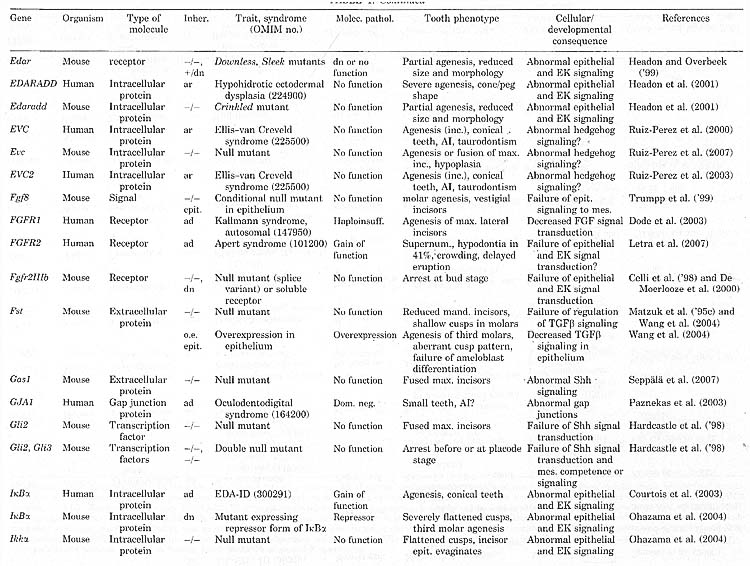
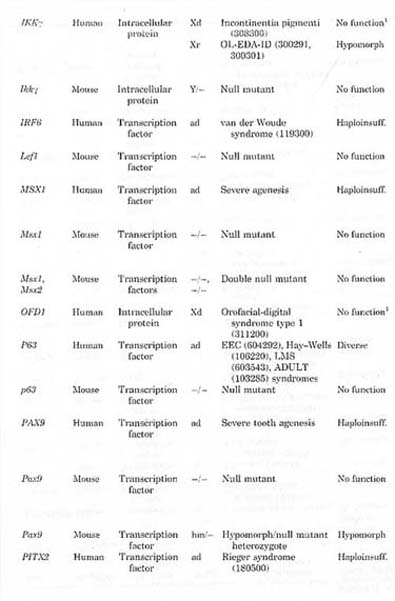
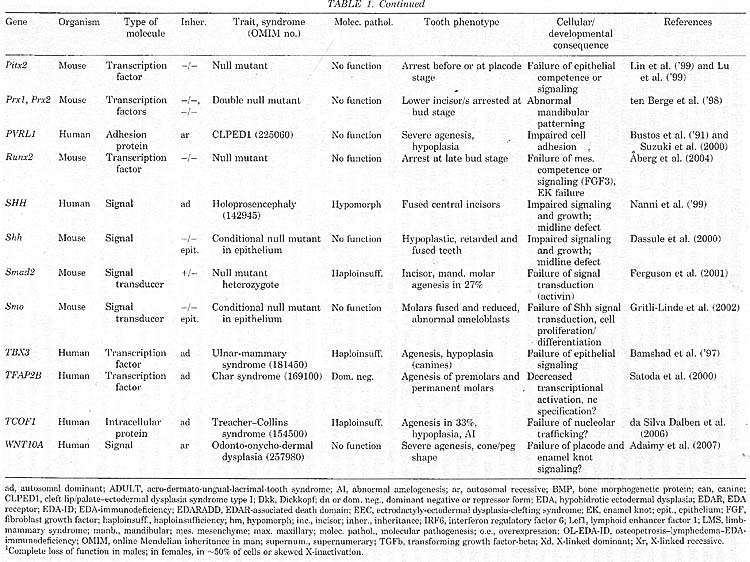
Молекулярная генетика началась с идентификации генетических причин агенеза зубов (Table 1). Так что основной прогресс был достигнут в агенезе зубов, ассоциированном с синдромами и несиндромным (isolated) семейным тяжелым агенезом зубов (oligodontia). Последний включает дефекты в MSX.l, PAX9, AXIN2 и EDA. Синдромы включают, помимо прочего, эктодермальные дисплазии и синдромы орального расщепления, а идентификация мутаций выявила общие генетическое пути для развития зубов и др. эктодермальных органов, а также зубов и черепно-лицевых структур.
В целом идентифицированные гены, которые затрагивались при разных формах агенеза зубов, оказались почти исключительно генами, которые регулируют развитие и их функции уже были изучены у мышей. Идентифицированные у человека и мыши гены, мутации которых затрагивали развитие зубов, касались всех основных сигнальных путей и транскрипционных факторов, обеспечивающих эти сигналы. Новые находки функций генов, которые мутантны при определенных синдромах у человека, ещё подкрепили эту тему (Ferrante et ah, 2006; Adaimy et al., 2007; Ruiz-Perez et al., 2007). Однако мутации в структурных и метаболических генах также могут приводить к нарушению развития зубов, как , напр., при несовершенном остеогенезе (osteogenesis imperfecta), diastrophic dysplasia и синдроме ectodermal dysplasia CLPED1 (Table 1).
MSX1 и PAX9 IN TOOTH AGENESIS
Первыми генами, идентифицированными при несиндромном агенезе зубов, были гены транскрипционных факторов MSX1 и PAX9, принадлежащие к гомеобоксным и paired-боксным семействам, соотв. (Vastardis et al., '96; Stockton et al., 2000). Как первоначально было показано в исследованиях на мышах, оба гена необходимы для развития зубов (Satokata и Maas, '94; Peters и Balling, '99). Вплоть до сегодняшнего дня было опубликовано большое количество мутаций в PAX9 и MSX1 в связи с тяжелым доминантным агенезом зубов (Figs. ID и 3). Наша лаб. участвовала в идентификации 5 мутаций в PAX9 и трех в MSX1 (Nieminen et al., 2001; Lammi et al., 2003; Klein et al., 2005; Tallon-Walton et al., 2007, unpublished). Мы также показали, что делеции в коротком плече хромосомы 4, которые вызывают синдром Wolf-Hirschhorn, захватывают локус MSX1, если у пациентов присутствует тяжелый агенез зубов (Nieminen et al., 2003). Идентифицированные генные дефекты MSX1 и PAX9 включают делеции, nonsense, сдвига рамки считывания, а также missense мутации и последние в большинстве своем располагаются в ДНК-связывающих доменах (Fig. 3).
Сходство фенотипов частичного агенеза зубов в результате делеций и др. типа мутаций указывает на то, что мутации вызывают потерю функции одного из аллелей (Das et al., 2002; Nieminen et al., 2003; Devos et al., 2006). Это подтверждается экспериментальными доказательствами, показывающими, что мутантный белок неспособен соединяться с известными последовательностями ДНК мишени или активировать транскрипцию (Hu et al., '98; Jumlongras et al., 2004; Mensah et al., 2004; Kapadia et al., 2006). Все дефекты, связанные с созданием стоп-кодонов в экзоне 1 в MSX1 и экзоне 2 РАХ9, могут вызывать в мРНК, несущих мутацию, чувствительность к деградации за счет механизма распада. вызываемого nonsense мутациями, но это экспериментально было подтверждено только для мутаций, затрагивающих exon 2-intron 2 соединение PAX9
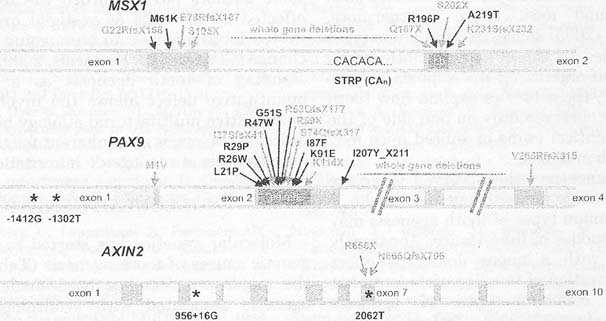 Fig. 3. Genomic structures и known mutations in MSX1, PAX9 и AXIN2. Mutations are shown above the schemes. Deletions и truncation mutations are with gray, missense mutations with black. Reported risk variants are shown below the schemes и with asterisks. Standard amino acid codes are used in the description of mutations. X, appearance of a stop codon (nonsense or frameshift mutations); fs, a frameshift as a result of an insertion or a deletion; light gray, exons; medium gray, coding region; dark gray, code for the DNA-binding domain; hb, homeobox.
Fig. 3. Genomic structures и known mutations in MSX1, PAX9 и AXIN2. Mutations are shown above the schemes. Deletions и truncation mutations are with gray, missense mutations with black. Reported risk variants are shown below the schemes и with asterisks. Standard amino acid codes are used in the description of mutations. X, appearance of a stop codon (nonsense or frameshift mutations); fs, a frameshift as a result of an insertion or a deletion; light gray, exons; medium gray, coding region; dark gray, code for the DNA-binding domain; hb, homeobox.
(Mostowska et al., 2005). Эти результаты строго указывают на то, что принципиальным эффектом мутаций является снижение количества функционального белка, демонстрируя гаплонедостаточность этих генов для развития зубов.
Итак, фенотипы, ассоциированные с мутациями
MSX1 и PAX9 показывают, что даже при существенной изменчивости в отдельных семьях и у отдельных пациентов, существуют типичные и отличающиеся паттерны для каждого гена (Kim et al., 2006; Nieminen, 2007) (Fig. 4). Хотя оба гена универсально затрагивают третьи моляры, достоверно более высокая частота агенеза обнаруживается во вторых премолярах и в верхнечелюстных первых премолярах в связи с мутациями
MSX1, чем с мутациями в
PAX9. С др. стороны, агенез верхнечелюстных первого и второго моляров и нижнечелючтных вторых моляров, достоверно более распространен в связи с дефектами в
PAX9. Обычно верхние боковые или нижние резцы также затрагиваются. Интересно, что некоторые сообщения о мутациях MSX1 описывают агенез первых постоянных моляров, даже если вторые моляры были развиты. Агенез молочных зубов был описан лишь в немногих случаях с мутациями
PAX9 (Nieminen et al., 2001; Das et al., 2002; Klein et al., 2005)
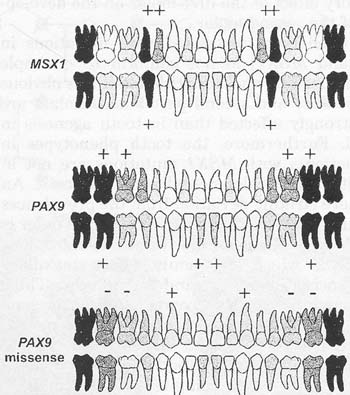 Fig. 4. Comparison of phenotypes associated with gene defects in MSX1 и PAX9. Only permanent teeth are shown. Darkness of the color expresses the frequency of agenesis. "PAX9 missense" includes only missense defects, i.e. single amino acid changes. agenesis significantly more frequent; -, agenesis significantly less prevalent.
Fig. 4. Comparison of phenotypes associated with gene defects in MSX1 и PAX9. Only permanent teeth are shown. Darkness of the color expresses the frequency of agenesis. "PAX9 missense" includes only missense defects, i.e. single amino acid changes. agenesis significantly more frequent; -, agenesis significantly less prevalent.
во всех остальных они выглядели в основном неповрежденными. Даже об изменениях размеров и морфологии пораженных зубов обсуждается лишь в немногих сообщениях, уменьшение размеров, укорочение корней и упрощение формы, по-видимому, довольно часто ассоциируют с мутациями
MSX1 и PAX9 (Jumlongras et al., 2001, 2004; Nieminen et al., 2001; Lammi et al., 2003; Klein et al., 2005). Боле того, в отдельных семьях описывается дисплазия ногтей и пациенты с оральными расщеплениями в ассоциации с агенезом зубов, вызываемым дефектами
MSX1 (van den Boogaard et al., 2000; Jumlongras et al., 2001). Несколько др. вариантов последовательностей
MSX1 были также обнаружены у пациентов с оральными расщеплениями, но вызможные аномалии зубов при этом не были описаны (Jezewski et al., 2003; Suzuki et al., 2004). С др. стороны, оральные расщепления не обнаруживались в всязи с гаплонедостаточностью MSX1 у пациентов с синдромом Wolf-Hirschhorn (Nieminen et al., 2003).
В случае PAX9, делеции всего гена и и мутации стартового кодона вызывали самые тяжелые дефекты, захватывающие премоляры и первичные моляры, т.е. нарушая образование всех postcanine зубов (Das et al., 2002; Klein et a!., 2005). С др. стороны, фенотипы missense дефектов были менее тяжелыми и напр., часто оставляли первые постоянные моляры незатронутыми (Fig. 4). Эти различия могут быть объяснены предположением, что в случае nonsense, frameshift и особенно missense дефектов некоторая функциональная активность белка сохраняется, возможно благодаря остаточной способности связывать ДНК и особенно благодаря межбелковым взаимодействиям.
Помимо мутаций
MSX1 и PAX9, многие синдромальные формы агенеза зубов вызываются доминантно действующими мутациями потери функции (Table 1 и ссылки). Эти мутации выявляют гаплонедостаточность развития зубов для нескольких генов, однако, с разными фенотипами агенеза. Сюда входят мутации в
PITX2 (Rieger syndrome),
IRF6 (van der Woude syndrome) и
p63 (EEC syndrome).
PATHOGENESIS OF MSX1 и PAX9 HAPLOINSUFFICIENCY
У мышей MSX1 и Pax9 экспрессируются в мезенхиме зубов, индуцируемые эпителиальными сигналами и участвующие в реципрокной передаче сигналов от мезенхимы к эпителию (rev. Peters и Balling, '99; Jernvall и Thesleff, 2000). У MSX1 и Pax9 нулевых мутантных мышей кондесация зубной мезенхимынарушена и развитие зубов блокировано на стадии почки (Satokata и Maas, '94; Peters et al., '98). Арест безусловно вызывается неспособностью мезенхимы индуцировать эпителиальный сигнальный центр, эмалевый узелок (knot), который необходим для перехода от стадии почки к ст. шапочки (SatoKata и Maas, '94; Peters et al., '98; Peters и Balling, '99; rev. Jernvall и Thesleff, 2000). Не очевидно, однако, как роль MSX1 и PAX9 в индукции эмалевого узелка связана с избирательным агенезом зубов, который затрагивает определенные зубы при образовании постоянных зубов, т.e. вторичные зубы и постоянные моляры, и обычно оставляет молочные зубы не затронутыми. Более позднее развитие и особенно зубов со можественными бугорками может быть более чувствительным к нарушению функции эмалевого узелка. Эмалевый узелок может также участвовать в регуляции, приводящей к формированию вторичных зубов. Онтогенетические и молекулярные механизмы образования вторичных зубов пока плохо изучены и стали предметом исследований лишь недавно (Fraser et al., 2006; Jarvinen et al., 2008, 2009).
В противоположность пациентам с гетерозиготной потерей функции при мутациях MSX1 или PAX9, мышиные гетерозиготы п нулевым мутациям не обнаруживают очевидного аномального феноипа. Однако снижение дозы Pax9 напр., в комбинации с гомпоморфным аллелем вызывает фенотип частичного агенеза зубов (Kist et al., 2005). У этих мышей, особенно ниженчелючстные резцы и задние моляры оказыаются повреждены. Сзади кпереди распоостранение агенеза моляров варьирует, но зависит от степени редукции дозы Pax9 и поэтому более задние моляры обнаруживают более высокую чувствительность к снижению дозы одиночного гена. Очевидно, что развитие различных моляров останавливается на разных стадиях развития зубов мыши и задерживается развитие всех моляров. Более того, кличество конденсированных зубных мезенхимных клеток снижено, вообще-то указывает на уменьшение коли чества детерминированных клеток (Kist et al., 2005). Однако неизвестно, действительно ли и как задержка развития или уменьшение конденсации причинно связаны с агенезом задних моляров. Зависимость развития моляров мыши от дозы Pax9 аналогична эффекту различных мутаций у человека, где полная инактивация одной копии PAX9 может затрагивать образование всех postcanine зубов, включая молочные моляры, тогда как др. типа мутации вызывают менее тяжелые фенотипы (Klein et al., 2005).
Т.к. Pax9, по-видимому, расположен выше MSX1 и действует синергично с ним (Peters et al., '98; Ogawa et al., 2006), то вполне возможно, что эффект гаплонедостаточности PAX9, по крайней мере, частично обусловлен снижением активности MSX1. Однако различия в фенотипах отражают независимые функции PAX9 и MSX1. Во время инициальных стадий развития зубов мыши, MSX1 преимущественно экспрессируется в области презумптивных резцов (rev. Tucker и Sharpe, 2004). Снижение инициально низкой экспрессии может сделать задние зубы более чувствительными к нарушению развития. Часто описываемый агенез нижнечелюстных первых постоянных моляров в присутствии вторых моляров у пациентов с мутациями MSX1 напоминает описания т. наз. 9-летних моляров вместо нормально развивающихся первых и вторых постоянных моляров (Rasmussen, '99). Если эти наблюдения действительно отражают неспособность к развитию первых постоянных моляров на неизвестной стадии, то это трудно понять в терминах простого количественного дефекта. Возможным объяснением может быть наблюдение ингибирующего эффекта, осуществляемого молярами на развитие более задних зубов (Kavanagh et al., 2007). Если снижение дозы MSX1 критически нарушает развитие первого моляра, оно может в то же самое время снижать ингибирующий эффект первого моляра на развитие второго моляра.
Фенотипы, ассоциированные с мутациями в
PAX9 и MSX1 не подтверждают простого общего эффекта на формирование всех зубов, но очевидно, что первый и второй постоянные моляры затрагиваются сильнее, чем агенез зубов в целом. Более того, фенотипы зубов у некоторых пациентов с мутациями MSX1 не согласуются онтогенетической последовательностью зубов. Наиболее поразительное отклонение от распространения на уровне популяций и последовательности развития наблюдается при синдроме Rieger, вызываемого мутациями в
PITX2, при котором обычно затрагиваются верхнечелюстные центральные резцы (Childers и Wright, '86). Т.о., даже благодаря количественному уменьшению дозы гена вызывается частичный агенез, фенотип, который не может быть объяснен просто общим уменьшением величины зуб-формирующего потенциала, затрагивающего последние развивающиеся зубы. В зависимости от гена, разные классы зубов, могут обнаруживать разную чувствительность. Эти чувствительности могут базироваться на дифференциальной экспрессии генов в разных классах зубов или на специфической роли в детерминации классов зубов (rev. Tucker и Sharpe, 2004; see also Mitsiadis и Smith, 2006; gene expression in tooth, http://bite-it.helsinki.fi). Дополнительные механизмы могут также участвовать в таких стимулирующих и ингибирующих взаимодействиях между развивающимися зубами и аномалиями в развитии лицевых отростков и черепно-лицевых тканях,окружающих зубы.
AXIN2 and PARADOXES OF WNT SIGNALING
Третьим геном, дефекты которого вызывают агенез зубов в качестве единственного порока развития, является
AXIN2, внутриклеточный агонист передачи сигналов Wnt. После исключения MSX1 и PAX9 и и проведения поиска по всему геному в финских семьях с тяжелым, но варьирующим агенезом зубов, мы идентифицировали nonsense мутацию потери функции в
AXIN2 на хромосоме 17 (Lammi et al, 2004) (Figs. 3 и 5). Фенотипы в этих семьях были поразительными в смысле тог, что первичные зубы были интактными даже у пациентов с наиболее тяжелым агенезом, у которых формировалось только три постоянных зуба. Это указывает на то, что функция AXIN2
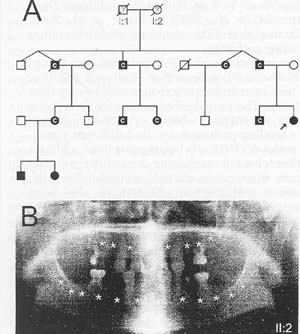 Fig. 5. AXIN2: pedigree и oligodontia phenotype. (A) Pedigree showing autosomal dominant inheritance of severe permanent tooth agenesis и colorectal neoplasia. Filled symbols, tooth agenesis; C, colorectal neoplasia. (B) Panoramic tomograph of 11:2 at age 34. Asterisks indicate congeni-tally missing permanent teeth. Many deciduous teeth persist.
Fig. 5. AXIN2: pedigree и oligodontia phenotype. (A) Pedigree showing autosomal dominant inheritance of severe permanent tooth agenesis и colorectal neoplasia. Filled symbols, tooth agenesis; C, colorectal neoplasia. (B) Panoramic tomograph of 11:2 at age 34. Asterisks indicate congeni-tally missing permanent teeth. Many deciduous teeth persist.
особенно важна для процесса, связанного с замещением зубов и развитием постоянных моляров. Более того, мутации также предрасполагали пациентов к colorectal неоплазиям, создавая тем самым необычную ассоциацию между агенезом зубов и раком.
Передача сигналов Wnt существенна для развития зубов, как показывают эксперименты с трансгенными мышами (rev. Mikkola и Millar, 2006). Эктопическая экспрессия внеклеточного Wnt антагониста
Dkkl в ротовом эпителии под контролем промотора keratin 14 вызывает арест развития на плакодной стадии, тогда как нулевые мутанты
Lefl, транскрипционного фактора, обеспечивающего передачу сигналов Wnt, арест развития зубов происходит на стадии почки (bud) (van Genderen et al., '94; Andl et al., 2002). Мутации потери функции
WNT10A ответственны за синдром рецесситвной odonto-onycho-dermal dysplasia с тяжелым агенезом зубов (Adaimy et al., 2007). С др. стооны, эктопическая экспрессия Lefl или избыточные уровни β-catenin в ротовом эпителии ведут к эктопическому развитю зубов (Zhou et al., '95; Jarvinen et al., 2006; Kuraguchi et al., 2006). Дерегуляция уровней β-catenin вследствие мутаций потери функции в adenomatous polyposis coll (APC) ведет к развитию челюстных osteomas, odontomas и добавочных зубов, также у человека (Gardner, '62; Wolf et al., '86). APC и AXINs являются компонентами внутриклеточного белкового комплекса, который противодействует передаче сигналов Wnt путем способствования деградации β-catenin, внутриклеточного мессенджера передачи сигналов Wnt, поэтому при мутациях потеря функции AXIN2, т.e. агенез зубов, наблюдается в точности наоборот тому, что ожидалось на основании находок др. исследований. Действие AXIN2 как регулятора негативной обратной связи указывает на то, что он необходим на определенных стадиях развития зубов, чтобы подавлять активность β-catenin. Более того, у условных мутантных мышей стимулирование передачи сигналов Wnt направляется на эпителий, тогда как
Axin2 экспрессируется также в мезенхиме. Возможно, что в ротовой и зубной мезенхиме следствия избыточной активации β-catenin могут быть совершенно иными, чем те, что в эпителии. Нулевые
Axin2 мутантные мыши дают фенотип краниосиностоза в результате повышенной пролиферации и дифференцировки потенциальных остеогенных клеток черепа (Yu et al., 2005), но неясно, встречается ли этот фенотип, связанный с агенезом зубов, у человека.
GENETICS OF TOOTH AGENESIS
Связь между агенезом зубов и предрасположеностью к раку как следствие дефектов AXIN2 объясняется регуляторной ролью передачи сигналов Wnt в развитии зубов (van Genderen et ah, '94; Jarvinen et al., 2006) и основным вкладом дерегуляции уровне внутриклеточного β-catenin в канцерогенез (Giles et al., 2003; Segditsas и Tomlinson, 2006). Интригует рассмотреть более общую взаимосвязь между агенезом зубов и предрасположеноостью к раку и тем, что агенез зубов может в некоторых случаях служить показателем последнего. Интересно, что повышенная распространенность агенеза зубов недавно была описана среди пациентов с овариальной эпителиальной карциномой (Chalothorn et al., 2008). Основной наследственный вклад в рак может вноситься в основном неизвестными аллелями с низкой пенетрантностью (Amundadottir et al., 2004; de la Chapelle, 2004), и некоторые из этих аллелей могут также вносить вклад в фоновый агенез зубов.Вполне возможно, что дефекты в др. генах, в дополнение к AXIN2, участвуют в передаче сигналов Wnt и регуляция уровня β-catenin также может связывать агенез зубов с предрасположенностью к раку. Интересно, что экспрессия
MSX1 снижается в клетках рака шейки матки и MSX1 способен стабилизировать p53, хорошо известный опухолевый супрессорный белок (Park et al., 2005).
TOOTH AGENESIS IN ECTODERMAL DYSPLASIA SYNDROMES: MUTATIONS IN THE EDA SIGNALING PATHWAY
Зубы обладают общими мезанизмами развития с др. эктодермальными органами, включая инициацию эпителиальных плакод и реципрокные взаимодействия эпителиального и мезенхимного компонентов (rev. Mikkoia и Millar, 2006). Использование общих генетических путей создает основу для эктодермальных дисплазий, группы, состоящей приблизительно из 200 врожденных забеоеванаий, затрагивающей волосы. ногти, зубы и eccrine железы (rev. Itin и Fistarol, 2004). В дополнение к варьирующему вовлечению зубов в разные формы эктодермальных дисплазий, минорные эктодермальные аномалии, они часто наблюдаются в ассоциации с тяжелым агенезом зубов (Schalk-van der Weide, '92; Nordgarden et al., 2001; Pirinen et al., 2001).
Лежащие в основе дефекты генов были идентифицированы при некоторых формах эктодермальных дисплазий (Table 1), это помогает очистить классификацию этих болезней. Позиционное клонирование генов, мутантных при гипогидротических ectodermal dysplasias (EDAs), привело к идентификации нового сигнаьного пути, передачи сигналов EDA (rev. Mikkoia и Thesieff, 2003). Дефекты в EDA, EDAR, EDARADD, IKKy и их мышиных гомологах, т.e. в сигнальных лигандах, их рецепторах и внтуриклеточных медиаторах передачи сигналов, инактивируют путь. В этом случае молекулярный патогенез и фенотипы у пациентов людей и мутантов мыши сравнимы. У мутантных мышей резцы и третьи моляры обычно неспособны к развитию, а первые моляры гипопластичны, тогда как у пациентов с EDA, агенез, тяжелый как в отношении прорезывания, так и морфологии зубов, упрощен (Soderholm и Kaitila, '85; Pispa et al., '99; Lexner et al., 2007) (Fig. IE). Фенотипы мышей с нарушением или избыточной экспрессией передачи сигналов EDA указывает на то, что ранние дефекты эктодермальных плакод и в зубах эмалевых узелков лежат в основе эктодермальных дефектов у пациентов (Pispa et al., '99; Mustonen et al., 2004). Т.о., нарушение передачи сигналов на ранней стадии ведет к аномалиям, которые также присутствуют в молочных зубах. Неадексатное образование эмалевых узелков может также объяснить конические и клино-образные зубы. Недавние поски в микромассивах генов мишеней для пути EDA идентифицировали набор компонент в др. ключевых сигнальных путях, регулирующих развитие зубов, включая передачу сигналов Shh и ингибиторы пути BMP, CCN2 и follistatin, а также ингибитор Wnt сигнала Dkk4 (Pummila et al., 2007; Fliniaux et al. 2008), указывая тем самым, что передача сигналов EDA модулирует др. сигнальные пути.
Мутации пути EDA являются или X-хромосомными, рецессивными или доминантными негативными, т.e. ведут к полной инактивации сигнального пути. Частичный , хотя и тяжелый агенез зубов указывает на перекрывание функций сигнальных путей, т.e. разные сигнальные пути обладают перекрывающимися функциями, это добавляет дальнейший элемент, объясняющий, как дефекты разных генов могут вызывать частичнфй агенез зубов. Недавно missense мутации в EDA были также описаны в связи с X-сцепленным семейным тяжелым агенезом зубов при отсутствии ассоциированных дефектов в др. эктодермальных органах и в этих случаях вполне возможно, что мутации вызывают только частичную инактивацию функции белка (Tao et al., 2006; Tarpey et al., 2007; Li et al., 2008).
В дополнение к передаче сигналов EDA функциональнае перекрывание, по-видимому, объясняет фенотип частичного агенеза зубов и вдр. случаях с рецессивным наследованием, напр., при cleft lip/palate, ectodermal dysplasia (CLPED1, nonsense мутации в
PVRLl), odonto-onycho-dermal dysplasia (nonsense мутации в
WNT10A) и Ellis-van Creveld (EVC) синдром (мутации в
EVC и EVC2) (Table 1).
TOOTH AGENESIS—A CLOSED CASE?
До сих пор идентификация мутаций была успешной в ассоциации с мультиорганными синдромами и при несондромном тяжелдом агенезе, олигодонтии. Для последней инактивирующие мутации были идентифицированы или семьях с полностью пенетрантной олигодонтией, которая следовала доминантному наследованию или открыты de novo, доминантно действующие или Х-сцепленные мутации. Генетические дефекты, ассоциированные с синдромами обычно также обнаруживают строгий эффект на функцию гена, т.e. потерю функции, доминантный негативный эффект или возможно избыточность функции, но при разных синдромах эффект на развитие зубов может быть сильным или менее пенетрантным и изменчивым (Table 1). Несмотря на неполную пенетрантность агенеза зубов в послдеднем случае диагноз синдрома возможен из-за др. существенных характеристик. Т.о., общим для идентифицированных генных дефектов является то, что они в общем оказывают сильный эффект на фугкцию гена и были идентифицированы у пациентов с относительно ясным диагнозом и способом наследования.
В дополнение к мутациям, описанным выше, мы провели мутационный анализ генов кандидатов. включая MSX1, PAX9 и AXIN2 в некоторых др. семьях с доминантно наследуемым тяжелым агенезом с негативными результатами (unpublished). Мутации в важных регуляторных регионах могут остаться не обнаруженными, но маловероятно, что они могут объяснить все эти случаи. Хотя и преждевременно давать оценку относительного вклада MSX1, PAX9 или AXIN2 в доминантно наследуемый несиндромный тяжелый агенез зубов (see also Kapadia et al., 2007), вполне очевидно, что участвуют ещё не распознанные гены. Строгий локус кандидат в хромосоме 10 был локализован в крупной китайской родословной (Liu et al., 2001). также вполне возможно, что мутации будут обнаружены в специфичных для зубов регуляторных элементах известных онтогенетически важных генов.
Несмотря на несомненный прогресс описанный выше, большинство генетических причин агенеза зубов остается нераспознанным. Сюда входят заметные фракции тяжелого агенеза зубов и особенно те, что без доминантного наследования, т.к. случаи при которых тяжелый фенотип не присутствует у родителей или др. сленов семьи. Это может фактически составлять большинсво от всех тяжелых случаев. Более того, существуют только предварительные данные о генетических факторах, лежащих в основе широко распространенных типов агенеза зубов, т.к. гиподонтии резцов и премоляров агенеза третьего моляра. Вполне возможно, что последний может также вносить вклад в случаи тяжелого агенеза зубов без доминантного наследования. Доказательства для комбинированных эффектов генетических факторов предолставляются случаями тяжелого агенеза, при которых оба родителя имеют менее тяжелые аномалии зубов и описаниями рецессивного наследования (Schalk-van der Weide, '92; Lyngstadaas et al., '96; Ahmad et al., '98; Pirinen et al., 2001). Из-за высокой генетической гетерогенности агенеза зубов, по-видимому, значительно чаще обнаруживаются случаи комбинаций причинных аллелей двух или более генов, чем обнаружение рецессивного наследования. Рецессивное наследование наиболее вероятно в ситуациях кровесмешения или в популяциях со строгой вероятностью эффекта основателя (Ahmad et al., '98; Pirinen et al., 2001). Более того, некоторые случаи, по-видимому, спорадичного или недоминантного тяжелого агенеза зубов могут быть объяснены X-сцепленными мутациями напр.,. при EDA.
INCISOR и PREMOLAR HYPODONTIA
Очевидно, что варианты генов, в которых мутации уже были идентифицированы, при тяжелом или синдромном агенезе, являются заманчивыми кандидатами на роль причинных факторов гиподнтии резцов и премоляров (Vieira, 2003). Напр., мутации гетерозиготной потери функции в MSX1 и PAX9 вызывают тяжелый агенез зубов, вполне возможно, что варианты последовательностей с менее драматическими эффектами на экспрессию или функцию белка могут быть ответственны за распостраненные фенотипы гиподонтии. Этого типа варианты могут рассматриваться как гипоморфные (снижение активности вместо полной потери функции) или как "аллели риска" и они могут располагаться или в кодирующих или некодирующих регионах и влиять скорее на количество продуцируемого белка, чем на функцию белка. Фенотипы, вызываемые с помощью гипоморфных аллелей мышиного Pax9 иллюстрируют эффект такого типа аллельных вариантов (Kist et al., 2005).
Исследования с использованием подходов к генам кандидатам сообщали о выборках из трех популяций (Fig. 3). Vieira et al. (2004, 2007) использовали выборку из 116 ядерных семей из Бразилии для изучения передачи аллелей из разных полимофризмов в MSX1, TGFA, PAX9, IRF6 и FGFRI к зхатронутым детям. Эти исследования подтвердили ассоциации определенных аллелей этих генов с агенезом зубов и , более того, взаимодействия между аллелями в MSX1 гене, по-видимому, ассоциируют как с агенезом зубов, так и оральными расщеплениями (Suzuki et al., 2004; Vieira et al., 2004). В исследованиях методом случайного контроля с 102 пациентами из Бразилии аллели из двух сцепленных single nucleotide polymorphisms (SNPs) в 5' фланкирующей последовательности гена PAX9 наблюдались более часто у пациентов с агенезом зубов (Peres et al., 2005). Наиболее представленный аллель из одного SNPs затрагивает предсказанный сайт связывания семейства транскрипционных факторов Kruppel. Два SNP аллеля в AXIN2 также были чрезмерно предствлены в выборке из 55 пациентов из Польши (Mostowska et al., 2006). Ассоциированный аллель SNP в экзоне 7 изменяет предсказанный энхансер сплайсинга в предсказанный репрессор. Однако, особенно в др. случаях, в которых отсутсвует очевидное функциональное объяснение, ассоциированные аллели сами по себе могут не быть причинными, а скорее указывать на др. причинный вариант неравновесного сцепления. Было бы интерсно посмотреть, могут ли эти результаты быть подтверждены в дальнейших исследованиях и если возможно, то экспериментальными или дальнешими биоинформационными методами.
Мы отобрали др. типа подход для идентификации причинных, генетических факторов для гиподонтиии резцов и премоляров и использовали анализ сцепления в семьях с несколькими поколениями (Arte, 2001; Nieminen, 2007). В этих семьях агенез одного или нескольких вторых премоляров, нижнечелюстных центральных резцов или верхних боковых резцов, или в некоторых случаях, клино-образных верхних боковых резцов, следует без сомнения доминантному наследованию. В большинстве семей затрагиваются преимущественно или вторые премоляры или резцы.
После инициальных эксклюзивных результатов по многообещающим генам кандидатам (Nieminen et al., '95; Arte et al., '96), были расширены исследования до поиска по всему геному, чтобы идентифицировать причинные локусы (Nieminen, 2007; manuscript in preparation). При многоточечном анализе только относительно низкие показатели были получены, когда все семьи были учтены вместе, исходя генетической гомогенности. С др. стороны, в большинстве индивидуальных семей поиск идентифицировал несколько привлекательных локусов, где все поврежденные наследовали один и тот же гаплотип. Их локусы также обнаруживают некоторое перекрывание. Достоверным подтверждением сцепления была полученная в области хромосомы 18, которая не содержала известных генов. участвующих в агенезе зубов.
Эти результаты мультиточечного анализа всего генома приложимы также к известным генам кандидатам, проявляющихся или известными дефектами генов у человека или у мутантных мышей. Для MSX1, рекомбинацией затронутые индивиды были обнаружены во всех семьях, тогда как AXIN2, MSX2, РAХ9, IRF6 и TGFA не могут быть исключены в некоторых семьях. Результаты не подтверждают ни существование одного или основного локуса для гиподонтиии резцов и премоляров, ни участие известных генов кандидатов, особенно MSX1.С др. стороны, результаты лучше всего согласуются с существованием нескольких локусов, некоторые из которых могут содержать ранее известные гены кандидаты.
CONCLUDING REMARKS
Most types of tooth agenesis can be considered to be caused by quantitative mechanisms affecting especially those teeth that develop latest in their tooth classes. In terms of the classic morphogenetic theories of tooth development (Butler, '39; Osborn, '78), this can be regarded as exhaustion or change of the field strength or odontogenic potential. Historically, reduced size of the dental lamina or tooth germs have been considered as causes of agenesis (Griineberg, '51; Bailit, '75; Brook, '84). In modern thinking based on the molecular и genetic studies of tooth development, tooth agenesis is a consequence of a qualitatively or a quantitatively impaired function of genetic networks, which regulate tooth development (Thesleff, 2006). Understanding a regulatory failure as a cause of agenesis is in line with the classic concepts of morphogenetic fields и self-organizing cell populations. However, as shown by variable phe-notypes associated with loss of function mutations in different regulatory genes, quantitative defects of regulation do not necessarily affect only the last developing teeth, but, for example, affecting the balance of activators и inhibitors may lead to more complex phenotypes (Kavanagh et al., 2007).
Tooth size variation is limited to a certain range, apparently by regulative developmental mechanisms capable also to compensate for earlier deviations - (Glasstone, '63). Occurrence of truly microdontic teeth as observed after childhood cancer treatments (Holttii, 2005) appears to be rather rare (personal communications). Thus, if agenesis follows from a quantitative defect, it must somehow be converted to a qualitative change during development. Observations of regressing rudimentary tooth germs in wild-type animals as well as in mutant mice suggest that agenesis follows from failures to overcome critical developmental thresholds (Tureckova et al., '95; Mustonen et al., 2004) (Table 1). In several null mutant mice lines, tooth development is arrested at specific stages, и in mutants in which regulatory genes or pathways are manipulated quantitatively, supernumerary teeth develop from rudiments that are arrested in wild-type mice (Mustonen et al., 2004; Kassai et al., 2005; Klein et al., 2006). Apparently most critical are the stages of formation of the signaling centers that have an organizing role for the future development, in particular placodes и enamel knots. The other stages may include e.g. differentiation и activation of a program for secondary tooth formation. Overcoming the thresholds appears more critical in the late developing teeth. The reduction of the "tooth-forming potential" may follow from a reduced functional activity of a single gene as in the case of the heterozygous loss of function mutations or from a complete inactiva-tion of a single signaling pathway, the function of which is partially redundant as in the case of impaired EDA signaling. However, it is conceivable that a failure to overcome a developmental threshold may also follow from combined effects of several genetic factors.
All genes that have been associated with tooth agenesis (Table 1) have important developmental functions in many tissues и organs in addition to being necessary» for normal tooth formation. In fact, no regulatory genes are known at present, which would be necessary only for tooth development. Thus, it appears that a strict distinction between syndromic и nonsyndromic tooth agenesis may be relevant only regarding individual mutations и phenotypes that depend on the severity of the mutation. On the other hand, hypomorphic variants or risk alleles of genes in which defects cause severe tooth agenesis or syndromes are plausible candidates to underlie the common types of tooth agenesis.
For several reasons, new genes underlying different types of tooth agenesis will most probably be identified. Taken the complexity of development of human dentition и the regulatory genetic networks, it is conceivable that several genes involved in these genetic pathways may contribute to tooth agenesis. In particular, the results from our genome-wicle search suggest novel loci for incisor и premolar hypodontia. Thus, the background of tooth agenesis is most probably even more genetically heterogeneous than it appears to be generally assumed today.
Сайт создан в системе
uCoz
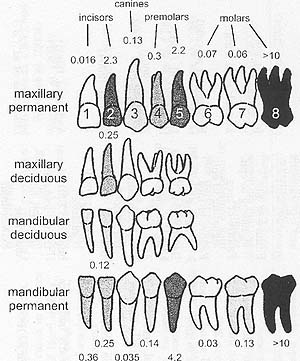 Fig. 2. Prevalences of agenesis of different teeth. Prevalences from teeth on the left и right are combined. Darkness of color indicates the prevalence и figures indicate prevalences as percentages. Figures inside the crowns of the maxillary permanent teeth are the second digits of the FD1, Federation Dentaire Internationale codes. Prevalences of permanent teeth except third molars are according to Table 4 in Polder et al. (2004).
Fig. 2. Prevalences of agenesis of different teeth. Prevalences from teeth on the left и right are combined. Darkness of color indicates the prevalence и figures indicate prevalences as percentages. Figures inside the crowns of the maxillary permanent teeth are the second digits of the FD1, Federation Dentaire Internationale codes. Prevalences of permanent teeth except third molars are according to Table 4 in Polder et al. (2004).