Посещений:  СЛИЯНИЕ МИОБЛАСТОВ
СЛИЯНИЕ МИОБЛАСТОВ
Сигнальные механизмы
|
|
Signaling Mechanisms in Mammalian Myoblast Fusion
Sajedah M. Hindi, Marjan M. Tajrishi and Ashok Kumar  Science Signaling v.6, (272), re2. [DOI: 10.1126/scisignal.2003832] (23 April 2013) |
Myoblast fusion is a critical process that contributes to the growth of muscle dur-
ing development and to the regeneration of myofi bers upon injury. Myoblasts fuse
with each other as well as with multinucleated myotubes to enlarge the myofi ber.
Initial studies demonstrated that myoblast fusion requires extracellular calcium
and changes in cell membrane topography and cytoskeletal organization. More
recent studies have identifi ed several cell-surface and intracellular proteins that
mediate myoblast fusion. Furthermore, emerging evidence suggests that myo-
blast fusion is also regulated by the activation of specifi c cell-signaling pathways
that lead to the expression of genes whose products are essential for the fusion
process and for modulating the activity of molecules that are involved in cytoskel-
etal rearrangement. Here, we review the roles of the major signaling pathways in
mammalian myoblast fusion.
|
 Группа миобластов с окрашенными пурпурным цветом микротрубочками, окрашенными красным - телами Гольджи и окрашенными голубым - ядрами Группа миобластов с окрашенными пурпурным цветом микротрубочками, окрашенными красным - телами Гольджи и окрашенными голубым - ядрами
Скелетные мышечные волокна являются синцитием, который возникает в результате слияния миобластов во время развития. У взрослых слияние миогенных клеток необходимо для облегчения роста и репарации мышечных волокон после повреждения. На клеточном уровне процесс слияния характеризуется выстраиванием миобластов и мембран мышечных трубок и перестройкой актинового цитоскелета в местах контакта, что сопровождается слиянием мембран, которое протекает в две стадии. Сначала слияние между миобластами (которое обозначается как "первичное слияние") приводит к образованию зарождающихся мышечных трубок. Во второй фазе миобласты сливаются с формирующимися мышечными трубками (в результате процесса, известного как "вторичное слияние"), которое приводит к добавлению ядер и росту мышечных трубок (1, 2). Наибольший прогресс в понимании процесса слияния миобластов во время развития достигнут в исследованиях на Drosophila melanogaster, у которых генетический скрининг идентифицировал участие специфического молекулярного аппарата (1, 2). Исследования последних десяти лет выявили несколько генов кандидатов, продукты которых могут действовать и у млекопитающих (1, 3, 4). Современное понимание слияния миобластов у млекопитающих базируется в основном на экспериментах с культурой миобластов, в которой воспроизводятся ступени слияния in vitro. Эти исследования показали, что слияние миобластов у млекопитающих регулируется различными белками клеточной адгезии, включая α3-, α9- и β-integrin субъединицы, neogenin, M- и N-cadherin, CD36 и disintegrin и metalloprotease12 (ADAM12); трансмембранные липиды, включая холестерол и phosphatidylserine; и внутриклеточные домен-ассоциированные сигнальные или адапторные белки, включая β-catenin, end binding 3 (EB3), kindlin-2, myoferlin, creatine kinase B, diacylglycerol kinase ξ, Rac1, focal adhesion kinase (FAK) и syntrophin, которые накапливаются в местах контакта между двумя миогенными клетками или симметричным или асимметричным способом (1, 3). Более того, сегодня установлено, что ряд клеточных сигнальных путей играет критическую роль в влиянии миобластов (Fig. 1). Некоторые из этих путей активируются в результате присоединения специфических белков клеточной поверхности между сливающимися партнерами, тогда как др. активируются как часть программы миогенной дифференцировки, но они вносят вклад в процесс слияния (Fig. 1). Мы рассмотрим роль основных сигнальных путей, участвующих в слиянии миобластов у млекопитающих.
Integrins and Focal Adhesion Kinase
Интегрины являются гетеродимерными белками, состоящими из α и β субъединиц, которые обнаруживаются в плазматической мембране клеток млекопитающих. Интегрины позволяют клеткам слипаться с внеклеточным матриксом и соединяться с различными внутриклеточными белками, соединяя клеточный экстерьер и интерьер. Генетические исследования показали, что β1 integrin субъединица в миогенных клетках является важным компонентом в первичном слиянии миобластов и сборке саркомеров у мышей (5). Одной из молекул, которая обеспечивает передачу сигналов integrin, является не рецепторный белок tyrosine kinase FAK (6). Соединение интегринов с лигандами внеклеточного матрикса - такими как fibronectin, vitronectin, vascular cell adhesion molecule 1 (VCAM1), collagen и laminin-вызывает образование кластеров интегринов и рекрутирование и аутофосфорилирование FAK и её ассоциацию с др. сигнальными белками, такими как Src, Cas и paxillin, которые ведут к активации нижестоящих сигнальных путей (6).
FAK участвует в слиянии миобластов in vitro и in vivo (7). Фосфорилирование FAK временно увеличивается во время миогенной дифференцировки, а ингибирование этого процесса блокирует слияние миобластов без нарушения экспрессии генов, связанных с дифференцировкой, это указывает на то, что FAK контролирует морфологические параметры миогенеза (7). Кроме того, целенаправленная делеция FAK в сателлитных клетках взрослых мышей ослабляет регенерацию миофибрилл в ответ на вызываемые BaCl2 повреждения.
Хотя не было отмечено различий в количестве регенерирующих миофибрилл в этом исследовании, но диаметр вновь сформированных волокон был существенно меньше у мышей со специфичной для сателлитных клеток делецией FAK спустя 5 дней после повреждения по сравнению с таковыми у мышей дикого типа, потенциально как результат снижения слияния миобластов (7). Хотя FAK, по-видимому, обеспечивает слияние миобластов in vitro и в ответ на повреждение мышц, необходимо установить участие FAK также в слиянии во время эмбриогенеза
Передача сигналов посредством FAK влияет на концентрацию некоторых молекул, которые участвуют в регуляции актинового цитоскелета, фокальной адгезии, Wnt, mitogen-activated protein kinase (MAPK) и инсулинового сигнального пути (7). Caveolin-3 и β1D-integrin являются одними из белков, которые участвуют в слиянии миобластов и чьё количество увеличивается в результате FAK-обеспечиваемой передачи сигналов. Интегриновая субъединица β1D стоит выше FAK, это указывает на то, что существует двунаправленная передача сигналов в пути β1D-integrin-FAK во время слияния миобластов (7). Точные механизмы, которые ведут к активации и рекрутированию FAK на интегрины, остаются загадкой; однако, этот процесс может использовать и др. вторичные мессенджеры . одним из таких кандидатов является protein kinase C (PKC) изоформа PKCΘ, которая довольно многочисленна в клетках миогенного клона. Индуцированная активация PKCΘ усиливает фосфорилирование FAK, количество caveolin-3 и β1D integrin, а также степень слияния миобластов (7, 8).
В дополнение к этой интерпретации с интегринами, FAK может также способствовать слиянию посредством своего взаимодействия с др. рецепторами клеточной поверхности, такими как neogenin (9). Neogenin (который кодируется Neo1) и его лиганды присутствуют в дорсальных частях сомитов и во время развития скелетных мышц у мышей (10). Netrin2 вызывает активацию FAK в культивируемых первичных миобластах neogenin-зависимым способом (10). Хотя скелетные мышцы развиваются нормально, мутации генной ловушки в локусе Neo1 ( Neo1Gt/Gt мыши) приводят к образованию тонких миофибрилл. Более того, фосфорилирование FAK существенно снижено в развивающихся мышцах Neo1Gt/Gt мышей по сравнению с мышами дикого типа, это указывает на то, что диада netrin-neogenin является физиологическим стимулом FAK во время развития мышц (10). Хотя эти исследования подтверждают, что передача сигналов как integrin, так и neogenin использует FAK, остается определить, активируется ли FAK независимо в ответ на integrin и neogenin или функциональная активация FAK нуждается во взаимодействии между этими двумя рецепторами. Функции FAK не законсервированы у позвоночных и беспозвоночных. DFak56 является дрозофилийным гомологом FAK млекопитающих . В отличие от FAK-дефицитных мышей, которые являются эмбриональными леталями (11), Drosophila, лишенные DFak56 жизнеспособны и плодовиты. Более того, DFak56 не нужен для функции integrin в клеточной адгезии, миграции или передаче сигналов in vivo (12).
Rho Guanosine Triphosphatases
Rho семейство guanosine triphosphatases (GTPases) играет критические роли в различных клеточных процессах, включая реорганизацию цитоскелета и активацию нижестоящих киназ. C2C12 это линия миобластных клеток мыши, которая быстро дифференцируется в мышечные трубки в условиях с низким содержанием сыворотки (13, 14). Эти клетки широко используются для изучения миогенеза in vitro, хотя доказательства указывают на то, что они могут не воспроизводить все ступени миогенеза, которые происходят in vivo во время развития или дифференцировки первичных миобластов in vitro. Исследования миобластов
C2C12 идентифицировали RhoA, Rac1 и Cdc42 в качестве важных регуляторов миогенеза (15-19). Эти GTPases функционируют путем модулирования активности нижестоящих киназ и транскрипционных факторов, таких как p38 MAPK, c-Jun N-terminal kinases (JNKs) и serum response factor (SRF) (15, 16, 19). Активность RhoA быстро и временно увеличивается в миобластах после их инкубации в среде для дифференцировки. Поскольку RhoA необходима для инициальной индукции миогенеза (20), то она д. быть деактивирована перед слиянием миобластов. Активная RhoA снижает стабильность и нарушает локализацию M-cadherin, молекулу клеточной адгезии, которая играет существенную роль в слиянии миобластов (21). RhoE (22) и Rho GTPase-активирующи1 белок GRAF1 (GTPase regulator associated with focal adhesion kinase-1) супрессирует активность RhoA в дифференцирующихся миобластах (23). Важность GRAF1 в ингибировании RhoA и слиянии миобластов in vivo была продемонстрирована в исследованиях на Xenopus laevis. Истощенные по GRAF1 эмбрионы обнаруживают повышенную активность RhoA и дефектный миофибриллогенез, что приводит к прогрессирующей мышечной дегенерации, нарушению подвижности и эмбриональной летальности (23). В противоположность ингибированию, обусловленному c помощью RhoA, др. члены семейства Rho GTPase -Rac1 и Cdc42-стимулируют аппарат слияния как in vitro, так и in vivo (15).У Drosophila, две близко родственные Rac GTPases, Rac1 и Rac2 (но не Cdc42), активируются в ответ на слипание клеток и действуют совместно, чтобы обеспечить слияние миобластов (2, 24); однако слияние миобластов у млекопитающих требует как Rac1, так и Cdc42 (25). Vasyutina et al. исследовали роль Rac1 и Cdc42 в процессе слияния в экспериментах на мышах со специфическим условным нокаутом в мышечных предшественниках (25). Анализ мышц у мышей на эмбриональный день 11.5 (E11.5) и E12.5 показал, что делеция любой из этих GTPases не оказывает какого-либо
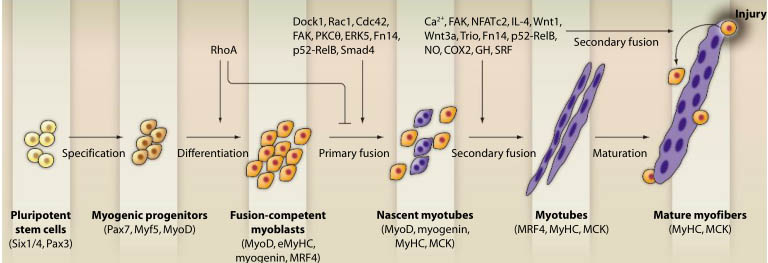 Fig. 1. The roles of different signaling molecules in primary and secondary myoblast fusion during myogenesis. Muscle progenitor
cells fi rst undergo myogenic commitment and differentiation to become fusion-competent myoblasts. The initial commitment to differentiation
requires the activity of the RhoA GTPase. Active RhoA interferes with myoblast fusion, and so it is deactivated before fusion occurs. A num-
ber of signaling molecules and pathways are activated in fusion-competent myoblasts that regulate primary myoblast fusion, which results
in nascent myotubes. Additional signaling molecules are then recruited, which lead to fusion of additional mononucleated myoblasts with
nascent myotubes. Secondary fusion also plays a critical role in the regeneration of injured myofi bers. Major signaling molecules involved in
primary and secondary myoblast fusion based on experimental evidence are depicted along the top. Specifi c myogenic markers expressed
at different stages in cells of myogenic lineage during myogenesis are noted along the bottom.
Fig. 1. The roles of different signaling molecules in primary and secondary myoblast fusion during myogenesis. Muscle progenitor
cells fi rst undergo myogenic commitment and differentiation to become fusion-competent myoblasts. The initial commitment to differentiation
requires the activity of the RhoA GTPase. Active RhoA interferes with myoblast fusion, and so it is deactivated before fusion occurs. A num-
ber of signaling molecules and pathways are activated in fusion-competent myoblasts that regulate primary myoblast fusion, which results
in nascent myotubes. Additional signaling molecules are then recruited, which lead to fusion of additional mononucleated myoblasts with
nascent myotubes. Secondary fusion also plays a critical role in the regeneration of injured myofi bers. Major signaling molecules involved in
primary and secondary myoblast fusion based on experimental evidence are depicted along the top. Specifi c myogenic markers expressed
at different stages in cells of myogenic lineage during myogenesis are noted along the bottom.
эффекта на миграцию, пролиферацию или дифференцировку клеток мышечных предшественников. Однако на более поздних стадиях (напр., на E14.5 и E18.5), развитие мышц нарушено, что проявляется в появлении коротких и тонких миофибрилл конечностей. Делеция генов, кодирующих или Rac1 или Cdc42 существенно снижает показатель слияния и количество ядер в развивающихся мышцах (25). Сходным образом культивируемые первичные миобласты от Rac1- или
Cdc42-дефицитных мышей обнаруживают заметное снижение способности к слиянию. Более того, активность Rac1 и Cdc42 необходима для обоих партнеров по слиянию (25). Хотя дефицит Rac1 или Cdc42 не влияет на сцепление или рекрутирование α- и β-catenin в места контактов между сливающимися партнерами, рекрутирование vinculin, F-actin и Vasp существенно уменьшается в места контактов. Особенно уменьшается рекрутирование Arp2/3, комплекса, индуцирующего полимеризацию актина, но только в Rac1-дефицитных миобластах, указывая тем самым, что Rac1 и Cdc42 могут обладать не перекрывающимися функциями (25). Одним из первых генов, идентифицированных как регулирующие слияние миобластов у Drosophila был mbc (26, 27), который кодирует guanine nucleotide exchange factor (GEF) , который де1йствует выше Rac GTPase в этом процессе (28). Myoblast city (MBC) является Drosophila ортологом белка млекопитающих Dock1. У млекопитающих, Dock2 и Dock5 являются дополнительными членами того же самого подсемейства Dock1-родственных белков (29). Хотя и Dock1
и Dock5 и их адапторный белок Crk и Crk-like 1 (Crkl) необходим для слияния миобластов быстрого типа у рыбок данио (30), Dock1 является главным GEF, чье отсутствие вызывает ингибирование развития скелетных мышц, из-за дефицита слияния первичных миобластов (31). Dock1-/- мыши погибают в течение минут после рождения из-за респираторной недостаточности и имеют диафрагму, которая сильно истончена (и обнаруживает нарушение прикрепления к межреберным мышцам) по сравнению с таковой дикого типа.
Сходное уменьшение массы отмечается в глубоких мышцах спины, языке и мышцах конечностей Dock1-/- мышей. Это уменьшение мышечной массы коррелирует с существенным снижением диаметра волокон на E18.5 у Dock1-/- мышей. Дальнейший анализ дефектов миогенеза у Dock1-/-мышей показал, что тяжелую цепь миозина (MyHC)-содержащие волокна выстраиваются один за др., но остаются одноядерными, это указывает на то, что Dock1 выполняет обязательную роль в слиянии первичных миобластов (31). Напротив, миотомы развиваются нормально у Dock5-/- и Dock1+/- мышей во время эмбриогенеза. Однако у Dock1-/-Dock5+/- мышей дифференцирующиеся миобласты остаются одноядерными, как и у Dock1-/- мышей, но обнаруживают дополнительные дефекты
в организации MyHC, элонгации и выстраивании клеток, указывая, что Dock5 играет важную роль в развитии миофибрилл на поздних стадиях (31). Эти находки также показали, что роль Dock1 законсервирована в сливающихся миобластах позвоночных и беспозвоночных. Предыдущее исследование показало, что Trio, двойной GEF для RhoA и Rac1, важен для для позднего эмбрионального развития и что он играет роль в формировании скелетных мышц (32). Однако миогенные дефекты у Trio-/- мышей отличаются от таковых у Dock1-/- мышей (31, 32). Кроме того, M-cadherin-зависимая адгезия активирует Rac1 посредством Trio во время слияния C2C12 миобластов (33). Эти находки открывают возможность, что хотя Dock1 необходим для слияния миобластов во время первичного миогенеза. Trio может играть роль во время вторичного миогенеза (Fig. 1). В противоположность своей роли у позвоночных, Trio γ не играет роль в слиянии миобластов во время развития Drosophila (24). Хотя необходимы дальнейшие работы, чтобы оценить полностью их роль, доступные, но имеющиеся доказательства подтверждают, что разные GEFs обеспечивают пространственную и временную регуляцию Rac1, это способствует слиянию миобластов во время эмбрионального развития и постнатального миогенеза посредством регуляции нижестоящих перестроек актинового цитоскелета в местах контакта между миобластами.
MAPKs
Пути передачи сигналов четырех MAPKs- extracellular signal-regulated kinase 1/2
(ERK1/2), JNK, p38 и ERK5-являются наиболее хорошо охарактеризованными клеточными сигнальными путями, которые активируются в ответ на различные внеклеточные стимулы и регулируют плюрипотентную клеточную функцию (34). Хотя стимулирующая роль p38 MAPK в миогенезе была установлена(35-37), как позитивная, так и негативная роли ERK1/2 и JNK были предположены (15, 38-40). Дальнейшие экспериментальные доказательства, касающиеся роли ERK1/2, JNK и p38 MAPK в слиянии миобластов, всё ещё отсутствуют. В раннем исследовании было продемонстрировано, что активность ERK5 возрастает в течение минут после воздействия на C2C12 миобласты среды с низким содержанием сыворотки и что она остается устойчивой во время терминальной дифференцировки (41).
Стимуляция ERK5 увеличивает активность E-box-содержащих промоторов, включая такие как Cdkn1a и Myl1 (41). Более того, ERK5 фосфорилирует белок детерминации миобластов 1 (MyoD) и myocyte enhancer factor 2C (Mef2C) in vitro, и это синергично увеличивает трансактивацию потенциала MyoD (41). Блокирование ERK5 c помощью экспрессии антисмысловых мРНК устраняет слияние миобластов и образование многоядерных мышечных трубок и у3меренно снижает экспрессию генов, связанных с дифференцировкой, таких как те, что кодируют MyoD, myogenin и p21 (41). Sunadome et al. оценивали роль ERK1/2 и ERK5 в слиянии миобластов и миогенной дифференцировке в экспериментах как с фармакологическими, так и молекулярными подходами (42). Специфическое ингибирование ERK1/2 не влияет ни на слияние, ни на дифференцировку C2C12 миобластов(42). Напротив, ERK5 является критической для слияния миобластов, но не для хода миогенной дифференцировки (42). Избыточная экспрессия доминантно негативной мутации ERK5 или действие short interfering RNA (siRNA)-обеспечивают нокдаун ERK5, существенно снижая степень слияния миобластов, приводя в результате к одноядерными миобластам, это указывает, что ERK5 важна для слияния первичных миобластов (42). Однако в противоположность предыдущему исследованию Dinev et al. (41), Sunadome et al. сообщили, что ингибирование ERK5 не оказывает эффекта на экспрессию мышце-специфических генов, таких как кодирующих MyoD, Mef2a, p21, MyHC и muscle creatine kinase, хотя наблюдается легкое снижение экспрессии генов, кодирующих myogenin и Mef2c на поздних стадиях дифференцировки (32). Эти наблюдаемые различия относительно экспрессии мышце-специфических генов в двух исследованиях можно приписать подходам, которые были использованы, чтобы ингибировать ERK5 (41, 42). Напр., возможно, что избыточная экспрессия ERK5-специфической антисмысловой мРНК, использованная в первом исследовании (31) также ведет к её связыванию с др. мРНК, которые обнаруживают гомологию с ERK5, это ведет к более выраженным эффектам на экспрессию мышце-специфических генов, чем это наблюдается с ERK5-специфической siRNA.
Изучены механизмы, с помощью которых ERK5 регулирует слияние миобластов. ERK5 способствует слиянию миобластов посредством активации нижестоящей транскрипции фактора specificity protein 1 (SP1), который, в свою очередь, соединяется с промоторами генов, кодирующих транскрипционные факторы Kruppel-like factor (Klf)2 и Klf4, приводя их к повышенной экспрессии (42). Количества как Klf2, так и and Klf4 увеличены в C2C12 миобластах после удаления сыворотки ERK5-зависимым способом, а ингибирование или избыточная экспрессия Klf2 и Klf4 влияют на слияние миобластов точно также как в случает ингибирования или избыточной экспрессии ERK5 (42). Заслуживает внимания, что повышенные активности ERK5 или Klf2 и Klf4 в отдельности недостаточны для индукции слияния миобластов; они способствуют слиянию миобластов в условиях, при которых миогенные транскрипционные факторы, такие как MyoD и Mef2C, активны и миобласты подвергаются дифференцировке (42). Nephronectin (который кодируется Npnt), белок внеклеточного матрикса, участвующий в слипчивости клеток с внеклеточным матриксом, ассоциирует с β1 интегриновой субъединицей в эмбриональных почках (43). Концентрация nephronectin возрастает во время миогенной дифференцировки и стимулирует слияние первичных миобластов (42). Промоторная область Npnt содержит множественные последовательности Klf2- и Klf4-связывающих сайтов и эти транскрипционные факторы индуцируют экспрессию Npnt во время слияния миобластов (42). Показано, что путь MEK5-ERK5-SP1-Klf2/4-Npnt является одним из наиболее охарактеризованных сигнальных путей, которые способствуют слиянию миобластов у млекопитающих (Fig. 2). Далее необходимо найти стимулы и исходные сигнальные события, приводящие к активации ERK5 во время миогенеза и определить, участвует ли этот путь также в слиянии миобластов во время эмбрионального развития и регенерации поврежденных мышечных волокон.
Calcineurin-NFATc2
Слияние миобластов в многоядерные мышечные трубки регулируется с помощью зависимой от кальция передачи сигналов (44-46). Среди нижестоящих мишеней зависимой от кальция передачи сигналов находится семейство NFAT транскрипционных факторов (NFATc1 to -c4), которое регулирует транскрипцию генов, чтобы скоординировать пролиферацию, жизнеспособность и дифференцировку в широком круге типов клеток (47, 48). Увеличение внутриклеточной концентрации кальция вызывает активацию сериновой и треониновой фосфатазы calcineurin, которая дефосфорилирует NFATc, делая возможной транслокацию в ядро NFATc и регуляцию транскрипции генов (48). Среди 4-х изоформ, NFATc2 стимулирует вторичное слияние миобластов (49). NFATc2 транслоцируется в ядро после первичного слияния и является важной для последующего добавления ядер в мышечное волокно. Хотя культивируемые первичные миобласты Nfatc2-/- мышей не обнаруживают заметных дефектов в своей экспрессии маркеров дифференцировки, их мышечные трубки меньшего размера и содержат меньше мышечных ядер. Дефекты слияния в Nfatc2-/- миобластах устраняются с помощью избыточной экспрессии NFATc2, это подтверждает, что он играет критическую роль в слиянии миобластов (49). В соответствии с ролью NFATc2 во вторичном слиянии, масса скелетных мышц и диаметр миофибрилл уменьшены у Nfatc2-/- мышей по сравнению с мышами дикого типа, без какого-либо изменения количества миофибрилл. Одним из механизмов, с помощью которого NFATc2 контролирует слияние миобластов, является тот, которые регулирует продукцию цитокина interleukin-4 (IL-4) (50). В то время как рецептор IL-4 (IL-4R) присутствует на всех этих стадиях дифференцировки в культурах миобластов, IL-4 продуцируется только после первичного слияния и NFATc2-зависимым способом (50). Поль IL-4 в слиянии миобластов также подтверждена находками, что диаметр миофибрилл меньше у мышей, дефицитных или по Il4 или Il4r, по сравнению с мышами дикого типа; такие мыши обнаруживают задержку мышечного роста после повреждения замораживанием , а миобласты от Il4-/- или Il4r-/- мышей имеют дефекты, сходные с теми, что обнаруживаются в Nfatc2-/- миобластах (50). Механизмы, с помощью которых IL-4 вызывает слияние миобластов изучены плохо; однако, исходя из его роли в др. типах клеток, таких как макрофаги, очень возможно, что IL-4 вызывает слияние миобластов за счет увеличения продукции молекул клеточной адгезии и с помощью индукции хемотаксиса (50). Др. молекула, регулируемая с помощью NFATc2-зависимых механизмов, это myoferlin (кодируемый Myof ), мембранный белок, существенный для слияния миобластов (51). Промотор Myof содержит множественные сайты связывания NFAT, а продукция myoferlin регулируется способом, сходным с таковым для NFATc2 в здоровых и поврежденных миофибриллах (51).
Помимо повышенных количеств кальция, некоторые др. стимулы могут активировать NFATc2 в сливающихся миобластах. Одним из таких факторов является prostaglandin F2α (PGF2α), который усиливает мышечный рост (52). В то время как PGF2α индуцирует образование мышечных трубок в IL-4-дефицитных клетках, он не усиливает продукцию IL-4 в миобластах дикого типа,
указывая тем самым, что NFATc2 может индуцировать слияние миобластов независимо от IL-4 (53). Более того, гормон роста (GH) также увеличивает активность NFATc2 в культивируемых миобластах (54). GH не вызывает гипертрофию в Nfatc2-/- мышечных трубках, это подтверждает роль NFATc2 в GH-опосредованной передаче сигналов в миогенных клетках (55). GH также индуцирует продукцию IL-4, это указывает на то, что GH может быть стимулом для активации пути NFATc2-IL-4 во время слияния миобластов (54).
Вторичное слияние миобластов также играет важную роль с мышечном росте и гипертрофии в ответ на упражнения с сопротивлением и механические нагрузки. Guerci et al. исследовали механизмы вызываемой перегрузками гипертрофии и открыли роль транскрипционного фактора SRF (56). Целенаправленная делеция Srf (который кодирует SRF) во взрослых миофибриллах (но не в сателлитных клетках) ингибирует перегрузками вызываемую гипертрофию и нарушает пролиферацию сателлитных клеток и их слияние с предсуществующими миофибриллами. SRF не нужен для активации передачи сигналов insulin growth factor-like 1 (IGF1)-Akt, которая обеспечивает зависимую от нагрузок гипертрофию. Напротив, SRF способствует пролиферации и слиянию миобластов паракринным способом. В частности, активация SRF в мышечных волокна в ответ на избыточные нагрузки ведет к повышенной продукции IL-6 и IL-4, которые усиливают пролиферацию сателлитных клеток и их слияние с предсуществующими миофибриллами. соотв. SRF не регулирует непосредственно экспрессию Il4; он, как полагают, усиливает экспрессию гена, кодирующего cyclooxygenase-2 (Cox2). Это исследование предоставило также первые доказательства, слияние сателлитных клеток является главным ограничивающим клеточным событием гипертрофии, вызываемой перегрузками (56).
NF-κB
Семейство NF-κB содержит 5 членов: RelA (также известен как p65), RelB, c-Rel, p105/p50 и p100/p52, которые дают гомо- и гетеродимеры (57, 58). В зависимости от типа стимула активация NF-κB происходит посредством канонического и неканонического сигнальных путей. Каноническая передача сигналов NF-κB связана с активацией ингибитора κB (IκB) kinase-β (IKKβ) и последующим фосфорилированием и деградацией IκB белка. Напротив, активация неканонического пути NF-κB нуждается в NF-κB-inducing kinase (NIK) и IKKβ, приводя к фосфорилированию и протеолитическому процессингу p100 субъединицы. чтобы сгенерировать p52 (57). Cellular inhibitor of apoptosis 1 (cIAP1) и cIAP2 являются одними из важных вышестоящих регуляторов как канонического, так и неканонического способов передачи сигналов NF-κB. cIAPs является также E3 ubiquitin лигазой, которая обеспечивает как Lys48 (K48)- так и (K63)-сцепленное убиквитинирование. В ответ на активирующие стимулы, адапторный белок tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) рекрутирует cIAPs на сигнальный комплекс, ассоциированный с рецептором, это вызывает K63-сцепленное убиквитинирование receptor-interacting kinase (RIP), приводя к активации нижестоящей IKKβ. cIAPs ингибирует также неканоническую ветвь NF-κB пути, облегчая K48-сцепленное убиквитинирование NIK, направляя его на протеосомную деградацию (59). При базовых условиях, cIAP, NIK, TRAF2 и TRAF3 формируют комплекс, который ведет к конституитивной деградации NIK посредством зависимого от протеосом пути. В ответ на активирующие стимулы комплекс TRAF2-TRAF3-cIAP помещается на
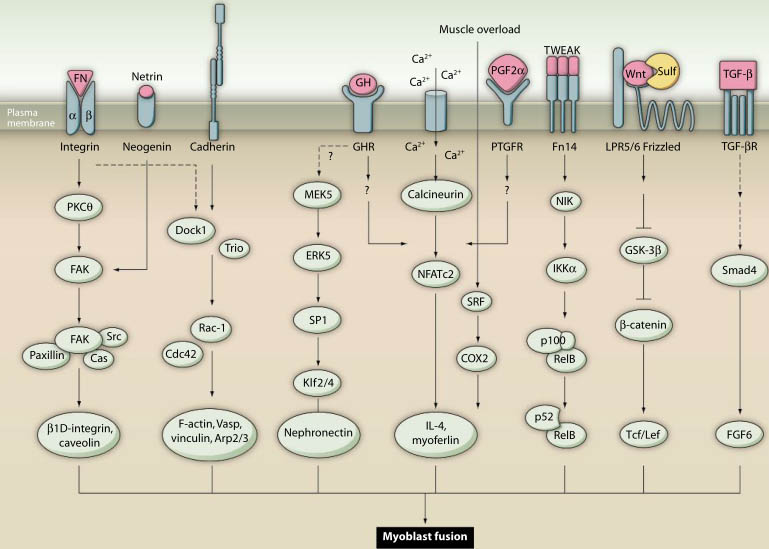 Fig. 2. The signaling mechanisms that stimulate myoblast fusion. Emphasis is placed on ligands and receptors that activate pathways, the main effectors of each pathway, and their target molecules.
Fig. 2. The signaling mechanisms that stimulate myoblast fusion. Emphasis is placed on ligands and receptors that activate pathways, the main effectors of each pathway, and their target molecules.
The interactions of integrins with their ligands, such as
fi bronectin (FN), causes the autophosphorylation of FAK by PKCΘ,
which leads to the activation of downstream signaling that results
in the increased expression of the genes encoding caveolin-3 and
the β1D integrin. Neogenin-netrin signaling also causes FAK activation. Ligation of M-cadherin activates Rac1 in a Trio- and Dock1-dependent manner. Cdc42 and Rac1 are major activators of vinculin, F-actin, Vasp, and the Arp2/3 complex for the cytoskeletal remodeling that occurs before myoblast fusion. The MEK5-dependent
activation of ERK5 promotes binding of the transcription factor SP1
to the promoter of the genes encoding the transcription factors Klf2
and Klf4, leading to their increased abundance. Subsequently, Klf2
and Klf4 bind to the Npnt promoter and induce the production of
nephronectin during myoblast fusion. Increases in intracellular cal-
cium concentration lead to activation of the serine-threonine protein
phosphatase calcineurin, which dephosphorylates and activates the
transcription factor NFATc2. Activated NFATc2 stimulates myoblast
fusion through the increased production of IL-4 and myoferlin. Possi-
ble stimuli for NFATc2 activation are PGF2α and GH. In response to
mechanical overload, the transcription factor SRF becomes activat-
ed, which leads to the increased abundance of IL-4 though COX2-
dependent mechanisms. Low amounts of the cytokine TWEAK
activate the noncanonical NF-κB pathway through activation of
NIK and IKKα. IKKα-mediated phosphorylation of p100 results in
its proteolytic processing to generate the p52 subunit and the sub-
sequent nuclear accumulation of p52-RelB heterodimers. Binding
of Wnt to Frizzled and LRP5/6 disrupts the binding of GSK-3β to
β-catenin, thus preventing the phosphorylation and degradation of
β-catenin. β-catenin then translocates to the nucleus and acts as a
transcriptional coactivator for Tcf/Lef target genes. Recruitment of
TGF-? to its cell-surface receptor leads to the activation of Smad4,
which translocates to the nucleus and recruits other transcription
factors to collectively induce the expression of target genes, such
as Fgf6. The dashed arrows indicate that direct interactions have
not yet been shown.
на рецептор, где TRAF2 обеспечивает K63-сцепленное убиквитинирование cIAP1 и
cIAP2, переключая его K48 ubiquitin лигазную активность с NIK на TRAF3. Деградация TRAF3 дестабилизирует комплекс TRAF-cIAP, тем самым делая возможным накопление вновь синтезируемой NIK и активацию нижестоящей IKKβ и последующее преобразование p100 в p52 (60-62). Несколько независимых исследований показали, что каноническая передача сигналов NF-κB ингибирует миогенную дифференцировку (58, 63); однако роль неканонической передачи сигналов NF-κB в формировании мышц привлекла мало внимания. Enwere с сотр. (64) сообщили, что неканоническая передача сигналов NF-κB способствует слиянию миобластов. Культивируемые миобласты от мышей, дефицитных по cIAP1 (который кодируется Birc2) обнаруживают повышенную способность к слиянию по сравнению с мышам дикого типа при инкубации в условиях низкого содержания сыворотки. Хотя делеция Birc2 усиливает слияние миобластов и активирует как канонический, так и неканонический пути, она ингибирует выход из клеточного цикла и экспрессию различных маркеров дифференцировки в культуре миобластов (64). Повышенное слияние миобластов приписывается дерепрессии неканонической передачи сигналов NF-κB, это также подтверждается находками, что siRNA-обеспечиваемое истощение p100, IKKβ bkb RelB-каждый из которых является компонентом неканонического пути-уменьшает добавления ядер и гипертрофию в культурах cIAP1-/- клеток (64). Напротив, избыточная экспрессия p52 и RelB усиливает первичное слияние миобластов в условиях низкого содержания сыворотки. Итак, эти находки подтверждают, что потеря cIAP1 вызывает активацию как канонического, так и неканонического NF-κB путей; первый ингибирует миогенную дифференцировку, тогда как второй способствует слиянию миобластов.
Bakkar et al. (63) ранее исследовали роль неканонической передачи сигналов NF-κB при миогенезе. Они сообщили, что активность канонического пути снижается во время дифференцировки C2C12 миобластов, с постепенным увеличением вклада неканонической передачи сигналов NF-κB посредством вышестоящей активации IKKα. В экспериментах с IKKα-/- и RelB-/- мышиными эмбриональными фибробластами (MEFs), авт. установили, что MyoD-индуцированная миогенная дифференцировка до некоторой степени редуцирована в этих клетках по сравнению с дикого типа MEFs. Однако, избыточная экспрессия IKKα не усиливает миогенную дифференцировку C2C12 миобластов или MyoD-экспрессирующих фибробластов. Эти до некоторой степени противоречивые находки двух исследований роли неканонического пути NF-κB в слиянии миобластов и миогенезе могут быть объяснены использованием разных экспериментальных условий. Исследования, осуществленные Bakkar et al. (63) использовали клеточные линии для некоторых экспериментов, которые могут вести себя по другому в отличие от первичных миобластов, которые были использованы Enwere et al. (64). В самом деле, первичные миобласты обладают существенно более высокой способностью к слиянию, чем C2C12 клетки, а небольшое и временное увеличение активности неканонической передачи сигналов NF-κB может быть достаточным, чтобы способствовать слиянию миобластов. Это также подтверждается находками, что фармакологическое ингибирование cIAP1 не усиливает слияние в культурах C2C12; вместо этого, оно ослабляет слияние, это ещё больше демонстрирует различия в механизмах слияния между первичными миобластами и C2C12 миобластами (64). Более того, Bakkar et al. не оценивали специфически слияние IKKα-избыточно экспрессирующих C2C12 миобластов или первичных миобластов от IKKα-/- мышей (63). В самом деле, не обнаруживаются фенотипические отклонения мышц у дикого типа и IKKα-/- мышей на перинатальной стадии (63), указывая тем самым, что роль некононической передачи сигналов NF-κB может быть компенсирована др. факторами во время эмбрионального развития скелетных мышц in vivo. Несмотря на это, специфическую роль IKKα необходимо исследовать далее в мышцах мышей IKKα-/- во время эмбрионального развития, а также слияние в культивируемых первичных миобластах от этих мышей. Сегодня доказано, что неканоническая передача сигналов NF-κB улучшает биогенез митохондрий в дифференцированных мышечных трубках благодаря индукции peroxisome proliferator-activated receptor-γ coactivator 1β (65).
Роль неканонического пути NF-κB в слиянии миобластов оценивали в исследованиях цитокина TNF-like weak inducer of apoptosis (TWEAK). TWEAK активирует как каноническую, так и неканоническую передачу сигналов NF-κB в разных типах клеток, включая миогенные клетки (66). Enwere et al. сообщили, что низкие концентрации TWEAK преимущественно активируют неканоническую передачу сигналов NF-κB и усиливает слияние культивируемых первичных миобластов (64). Эти находки согласуются с опубликованными сообщениями, что TWEAK рецептор Fn14 важен для образования мышечных трубок в культурах и для регенерации скелетных мышц взрослых в ответ на повреждения (67, 68). Ранее эффекты TWEAK на миогенез изучали на клеточной линии C2C12, которые показали, что в большом количестве TWEAK активирует канонический путь NF-κB и ингибирует миогенную дифференцировку (69). Хотя активация неканонического пути NF-κB специально не исследовалась в недифференцированных C2C12 миобластах, было также отмечено, что большие (но не малые количества) TWEAK активируют неканоническую передачу сигналов NF-κB в культурах дифференцированных C2C12 мышечных трубок (66, 70). Эти находки подтверждают, что линия клеток C2C12 и первичные миобласты мышей отвечают по-разному на одни и те же активирующие стимулы в отношении активации канонической и неканонической передачи сигналов NF-κB. Поскольку низкие дозы TWEAK могут усиливать слияние миобластов, то это заслуживает внимание поскольку продолжительное присутствие TWEAK может приводить к пагубным последствиям на возникающие мышечные трубки. Низкая концентрация TWEAK не только вызывает атрофию, его продолжительное присутствие может также снижать жизнеспособность дифференцированных мышечных трубок (70). Более того, регенерация мышц после вызванных cardiotoxin повреждений значительно снижается у TWEAK-трансгенных мышей, это потенциально является результатом хронического присутствия повышенных концентраций TWEAK у этих мышей
(71). Др. исследование показало, что спорадические inclusion-body myositis (IBM) мезангиобластов, которые дефектны по миогенной дифференцировке, продуцируют повышенные количества TWEAK в среде для дифференцировки и что нейтрализация TWEAK улучшает дифференцировку в культурах IBM мезангиобластов (72). Более того, связанная с мембраной форма TWEAK является более мощной, чем её растворимый вариант в отношении активации канонического пути NF-κB (73), это объясняет, почему трансгенная избыточная экспрессия или повышенная продукция связанного с мембраной TWEAK в болезненных условиях оказывает вредные эффекты на регенерацию мышц. Несмотря на этот недавний прогресс, механизмы, с помощью которых неканоническая передача сигналов NF-κB управляет слиянием миобластов, и её роль in vivo остаются неизвестными. Необходимы дальнейшие исследования для лучшего понимания роли неканонической передачи сигналов NF-κB в развитии скелетных мышц и регенерации миофибрилл в ответ на повреждения. Тем не менее доступная литература предоставляет первые доказательства, что активация неканонической передачи сигналов NF-κB улучшает здоровье мышц за счет усиления слияния миобластов и улучшения оксидативного метаболизма мышц.
Wnt Signaling
Wnt белки и секретируемые лиганды, которые передают свои сигналы через плазматическую мембрану за счет взаимодействия с Frizzled рецепторами и их ко-рецепторами low-density lipoprotein receptor-related protein 5 (LRP5) и LRP6 (74). После соединения со своими рецепторами Wnt белки индуцируют каскад внутриклеточных сигнальных событий, использующих белки, такие как Disheveled, axin, adenomatosis polyposis coli (APC) и glycogen synthase kinase-3β (GSK-3β), который достигает кульминации , стабилизируя β-catenin и его транслокацию в ядро, где он соединяется с T-cell factor/lymphoid enhancer-binding factor (Tcf/Lef), чтобы индуцировать транскрипцию генов мишеней для Wnt (74). В дополнение к этому каноническому пути определенные Wnt белки осуществляют свои эффекты с помощью активации пути planar cell polarity (PCP) (также известного как неканонический Wnt путь) и calcium/calmodulin-зависимого киназного пути (74, 75).
Передача сигналов Wnt важна как для развития эмбриональных мышц, так и для постнатального миогенеза (75). GSK-3β является критическим компонентом классического пути передачи сигналов Wnt, поскольку её инактивация ведет к стабилизации и ядерной транслокации β-catenin (75). Первоначальные доказательства роли передачи сигналов Wnt в слиянии миобластов были получены в исследовании Rochat с сотр. (76), в котором они установили, что ингибирование GSK-3β с помощью LiCl или совместного культивирования с Wnt1-экспрессирующими 3T3 фибробластами усиливает накопление в ядреβ-catenin и вызывает insulin-индуцированную дифференцировку покоящихся C2C12 миобластов (76). Добавление LiCl в среду спустя 36 - 48 ч после индукции дифференцировки приводит к заметному увеличению диаметра мышечных трубок, а также количества ядер в каждой мышечной трубке (76). Сходным образом, воздействие с помощью только Wnt1 достаточно для стимуляции слияния миогенных клеток (76). Было также сообщено, что воздействие др. Wnt лиганда, Wnt3a, увеличивает слияние C2C12 миобластов (77), далее было подтверждено, что классическая передача сигналов Wnt способствует слиянию культивируемых миобластов.
Роль канонической и неканонической передачи сигналов Wnt при слиянии миобластов in vivo установлена (78, 79). Каноническая передача сигналов Wnt индуцируется в клетках мышечных предшественников в течение 2 - 5 дней после повреждения мышц (79). Инъекция белка Wnt3a в поврежденную мышцу увеличивает размер регенерирующих миофибрилл без изменения количества миофибрилл, потенциально за счет слияния миобластов (79). Кроме того, Sulfs (6-O endosulfatases) активирует каноническую, но ингибирует неканоническую передачу сигналов Wnt в поврежденных миофибриллах (78). Sulf1 и Sulf2 избирательно удаляют 6-O-сульфатную группу с heparin sulfate (HS), это делает возможной HS proteoglycan (HSPG)-обеспечиваемую передачу сигналов. Sulf-обеспечиваемая HS 6-O-desulfation репрессирует передачу сигналов fibroblast growth factor (FGF) путём разрушения HS-FGF2-FGF receptor (FGFR) комплекса в сателлитных клетках, это делает возможной их дифференцировку, а, следовательно, образование новых миофибрилл (80). Мышце-специфическая делеция обоих Sulf1 и Sulf2 существенно задерживает образование миофибрилл после повреждений из-за дефектовs слияния миобластов (78). Сходным образом, культивируемые миобласты от мышце-специфических Sulf1 -/-Sulf2 -/- мышей обнаруживают снижение слияния после удаления FGF2 из культуральной среды. Это исследование также предоставило первые доказательства, что неканоническая передача сигналов Wnt активируется в ответ на повреждения мышц и что она ингибирует слияние миобластов (78). Так, Sulfs репрессирует неканоническую передачу сигналов Wnt во время регенерации мышц за счет усиления антагонистической канонической передачи сигналов Wnt (78). Т.к. Wnt3a, но не Wnt7a, содержит HS-связывающую Weintraub последовательность, то Sulfs может функционировать путем снижения степени соединения Wnt3a с HS, увеличивая доступ Wnt3a к активации канонической передаче сигналов Wnt. Кроме того, Sulfs стимулирует активацию и локализацию на мембране FAK во время слияния миобластов. Напротив, Wnt7a-обеспечиваемая неканоническая передача сигналов Wnt вызывает аберрантное субклеточное распределение FAK (78). Итак, эти данные предоставляют новую информацию относительно роли Sulfs и канонической и неканонической передачи сигналов Wnt при слиянии миобластов.
Other Signaling Pathways
Слияние миобластов также использует активацию ряда др. сигнальных молекул. Напр., передача сигналов канонического transforming growth factor-β (TGF-β) , которая функционирует посредством активации белка Smad4, ингибируя как пролиферацию, так и слияние C2C12 миобластов (81). Интересно, что Han et al. продемонстрировали, что целенаправленное удаление Smad4 ингибирует терминальную дифференцировку и слияние миогенных клеток во время развития языка путем ингибирования экспрессии Fgf6 (82). Воздействие экзогенного белка FGF6 частично восстанавливает слияние миобластов в Smad4-дефицитных первичных миобластах (82). Противоположные функции передачи сигналов TGF-β и Smad4 при слиянии миобластов, обнаруженные в этих двух исследованиях подтверждают, что др. члены сверхсемейства TGF-β также участвуют и что они могут функционировать путем регуляции отличающихся нижестоящих генов мишеней, чтобы контролировать слияние миобластов. Это может быть также объяснено различиями в механизмах слияния между первичными миобластами и C2C12 миобластами.
Nitric oxide (NO) продуцируется энзимом nitric oxide synthase в различных типах клеток, включая сосудистые эндотелиальные клетки, макрофаги, фибробласты, нейроны и поврежденные миофибриллы. NO является одним из критических мессенджеров, которые способствуют слиянию миобластов (83). Dahlman et al. далее показали, что во время развития концентрация NOS в стромальных фибробластах увеличивается благодаря NF-κB-зависимым механизмам, чтобы стимулировать слияние миобластов и мышечную гипертрофию (84).
Concluding Remarks
Our understanding of the process of myoblast fusion has taken a quantum jump in recent years. It is increasingly clear that the fusion process is a highly complex and elaborate network involving the coordination and crosstalk of many signaling pathways and that it involves both extracellular and intracellular events. These signaling molecules and transcription factors act in synergy or antagonism in feed-back and feed-forward loops. Future investigations should focus on identifying the extracellular stimuli that lead to the activation of signaling pathways that stimulate myoblast fusion and whether the activation of some of these involved pathways is perturbed in various muscle-degenerative disorders, such as muscular dystrophy. Furthermore, fusionrelated target genes regulated through the activation of these signaling pathways need to be identifi ed. Recent technological advances are paving the way for further investigation of the signaling networks that control myoblast fusion on a proteome- and
genome-wide scale.
|