Генетические дефекты пока могут быть идентифицированы только при тяжелых формах несиндромного агенеза зубов. Они затрагивают гомеобоксный ген
, ген внутриклеточного антагониста передачи сигналов Wnt (72). Все эти три гена, как было установлено, являются важными регуляторами ранних стадий развития зубов у мышей, особенно при переходе от стадии почки к стадии шапочки (50, 51). Следовательно, мутации, вызывающие oligodontia у человека, как полагают, являются результатом ареста развития зубов на стадии зачатка, когда гаплонедостаточность снижает дозу гена и, следовательно, потенциал развития зуба ниже критического уровня (51). Однако в противоположность этому можно ожидать, базируясь на подразумеваемой роли MSX1, PAX9 и AXIN2 в развитии зубов, олигодонтия, вызываемая дефектами этих генов, выявляет типичные, хотя и частично перекрывающиеся и сильно изменчивые паттерны агенеза зубов (Fig. 3b,c). Мутации
у человека не затрагивают все зубы одного и того же класса (56-68). Напр., мутации в гене
затрагивают в основном моляры. Мутации в
, по-видимому, дают комбинацию MSX1 и PAX9 фенотипов. Они затрагивают исключительно рост постоянных зубов, но никогда не затрагивают максиллярные центральные резцы (51, 72). Они, а также клыки и первые моляры являются очень стабильными зубами и редко обнаруживается врожденное их отсутствие (73). Наблюдение, что чувствительность к последствиям определенных генетических дефектов варьирует между первичными и постоянными зубами, также как и между классами и типами зубов вызывает некоторые вопросы относительно универсальной законности концепций контроля формирования зубов. В этом контексте, не нужно забывать, что наиболее распространенной моделью для изучения одонтогенеза, являются мыши, обладающие только одной генерацией и двумя классами зубов.
Генетические дефекты, ассоциированные с синдромальными формами гиподонтии, в основном затрагивают другие гены, чем гены, ассоциированные с не синдромальными формами олигодонтии. Исключением являются мутации MSX1, которые также могут вызывать синдром Witkop, при котором дефекты ногтей пальцев рук и ног сопровождают агенез зубов (74). Наиболее выраженные дефекты кожи и её придатков в комбинации с гиподонтией и уменьшением размеров зубов наблюдаются при некоторых формах эктодермальной дисплазии, которая вызывается генетическими дефектами EDA пути, т.e. генов EDA, EDAR, EDARADD, IKKy, NEMO и p63 (50, 51, 75-78). Интересно, что мутации в EDA могут быть также ответственны за не синдромальную гиподонтию (79, 80). Поразительный фенотип агенеза зубов характеризуется единообразным, чрезвычайно редким отсутствием максиллярных центральных резцов, наблюдаемым при синдроме Rieger, который вызывается дефектами гомеобоксного гена PITX2 (51, 77, 81).
Семейное проявление и конкордантность агенеза зубов у близнецов указывают на значительное генетическое влияние также при умеренных формах гиподонтии, напр., широко распространенный агенез премоляров-резцов (51, 82-85). Если олигодонтию рассматривать как следствие критической недостаточности дозы гена, то вполне разумно предположить, что умеренная гиподонтия является результатом вариантов последовательности ДНК, которые менее тяжело сказываются на функции гена. Однако исследования полиморфизмов генов кандидатов, ответственных за олигодонтию, дают противоречащие результаты (86-90). Существенную роль в гиподонтии, как было установлено, играют варианты и гаплотипы TGFA (86, 91), IRF6 (92, 93), FGFR1 (92) , а также MMP1 и MMP20 (94). Т.о., по-видимому, отсутствует главный локус гиподонтии (51).
Amelogenesis imperfecta
Термин amelogenesis imperfecta (AI) обозначает наследственное нарушение развития зубной эмали. В строгом смысле определение включает только дисплазии эмали, которые возникают в отсутствие дефектов в др. тканях. Современная широко используемая номенклатура AI базируется на способе наследования, фенотипе и (если известна) молекулярной причине дефекта эмали (95). Фенотипическая классификация учитывает, что эмаль образуется в два основных этапа. На первом этапе, секреторной стадии амелогенеза, амелобласты секретируют органический матрикс, в котором рыхло откладываются кристаллы hydrox-yapatite. На втором этапе большинство матричных белков деградирует и резорбируется амелобластами, тогда как кристаллы растут в толщину до тех пор, пока они не приходят в контакт др. с др. и плотность минерала в эмали не достигнет примерно 95% (96). Нарушения секреторной стадии вызывают гипопластического типа AI, характеризующуюся количественным дефицитом эмали (Fig. 4a-f). Количественный дефицит колеблется в пределах от (действительной) аплазии, наз. smooth hypoplastic AI (Fig. 4a,c,e), до вертикальных борозд (Fig. 4b), горизонтальных углублений или ямок (Fig. 4d,f), в целом обозначаемых как грубая (локальная) гипопластическая AI. Нарушения процесса минерализации приводят к гипоминерализованному типу AI, характеризующейся количественным дефицитом, т.е.
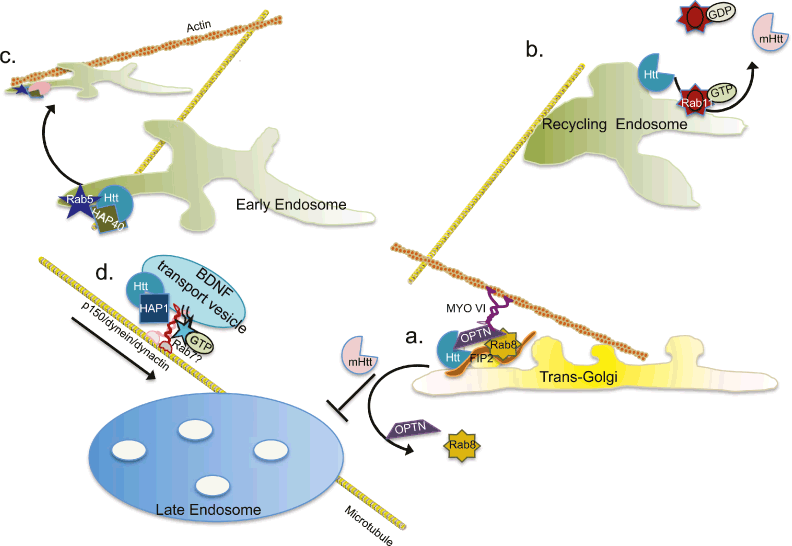 |
Fig. 4. Smooth (a, c, e) and rough (b, d, f) hypoplastic forms of amelogenesis imperfecta, (a) Intraoral view of the maxillary dentition of a female patient showing the conical shape of the tooth crowns, which in the absence of enamel is determined by the dentin cores (crowns of the incisors had been constructed prosthodontically). Note the missing right second molar (arrow), the eruption of which is delayed in comparison with the contralateral tooth, (c) Intraoral radiograph from the brother of the patient shown in (a): No enamel can be recognized, (e) Backscattered electron micrograph of an upper third molar surgically removed from the patient shown in (a): In comparison with the enamel (E) of a healthy third molar (g), the mineral density of the enamel-like material of the patient's tooth is essentially normal, but its thickness is only about 3-5%. (b) Frontal view of the incisors of a female patient showing vertica' enamel furrows and streaks (arrows), as they occur as a result of X-chromosome inactivation (Lyonization). How these furrows and streaks arise is illustrated by a light micrograph from an incisor tooth germ (h) at the stage of enamel (E) and dentin (D) formation: X-chromosome inactivation is a random process taking place in the stem cells, which reside in the cervical loop (CL). The result of inactivation is propagated to all descendant daughter cells that differentiate in the vertical direction (arrow) and produce new inner enamel epithelial (IEE) cells to finally become enamel-forming cells, ameloblasts. As a consequence, clusters of ameloblasts arise along the circumference of the tooth germ, which carry an X chromosome with either a normal or defective AMELX gene and alternatively produce a normal or defective enamel matrix, (d) Frontal view of the incisors of a female patient showing enamel pits, (f) A backscattered electron micrograph of a maxillary premolar from the same patient reveals similar pits associated with regions of slightly hypomineralized enamel (arrow), which account for the slight yellow-brown discoloration. D, dentin, DP, dental papilla, OEE, outer enamel epithelium. Original magnifications (e) xllO, (f, g) x50, (h) x20.
|
Fig. 4. Smooth (a, c, e) and rough (b, d, f) hypoplastic forms of amelogenesis imperfecta, (a) Intraoral view of the maxillary dentition of a female patient showing the conical shape of the tooth crowns, which in the absence of enamel is determined by the dentin cores (crowns of the incisors had been constructed prosthodontically). Note the missing right second molar (arrow), the eruption of which is delayed in comparison with the contralateral tooth, (c) Intraoral radiograph from the brother of the patient shown in (a): No enamel can be recognized, (e) Backscattered electron micrograph of an upper third molar surgically removed from the patient shown in (a): In comparison with the enamel (E) of a healthy third molar (g), the mineral density of the enamel-like material of the patient's tooth is essentially normal, but its thickness is only about 3-5%. (b) Frontal view of the incisors of a female patient showing vertica' enamel furrows and streaks (arrows), as they occur as a result of X-chromosome inactivation (Lyonization). How these furrows and streaks arise is illustrated by a light micrograph from an incisor tooth germ (h) at the stage of enamel (E) and dentin (D) formation: X-chromosome inactivation is a random process taking place in the stem cells, which reside in the cervical loop (CL). The result of inactivation is propagated to all descendant daughter cells that differentiate in the vertical direction (arrow) and produce new inner enamel epithelial (IEE) cells to finally become enamel-forming cells, ameloblasts. As a consequence, clusters of ameloblasts arise along the circumference of the tooth germ, which carry an X chromosome with either a normal or defective AMELX gene and alternatively produce a normal or defective enamel matrix, (d) Frontal view of the incisors of a female patient showing enamel pits, (f) A backscattered electron micrograph of a maxillary premolar from the same patient reveals similar pits associated with regions of slightly hypomineralized enamel (arrow), which account for the slight yellow-brown discoloration. D, dentin, DP, dental papilla, OEE, outer enamel epithelium. Original magnifications (e) xllO, (f, g) x50, (h) x20.
почти нормальной толщины, но уменьшено содержание минерала и повышено содержание белка (Fig. 5a-g). Умеренное снижение плотности минерала в основном ограничено границами между эмалевыми призмами (Fig. 5e), приводя к недозрелому типу AI (Fig. 5a,c,e). Результатом ещё более тяжелого дефицита минерала и удержания белка является гипокальцифицированная форма AI (Fig. 5b,d,f). Большинство генетических дефектов, отвечающих за AI, было идентифицировано в генах белков и энзимов эмалевого матрикса, необходимых для деградации эмалевого матрикса
 |
Fig. 5. Hypomaturated (a, c, e, g) and hypocalcified (b, d, f) forms of amelogenesis imperfecta, (a) Lateral view of the posterior permanent teeth of a female patient showing extensive enamel chipping, because hypomaturated enamel exhibits reduced resistance to mechanical loading, but is hard enough to break, (c) In the panoramic radiograph from the same patient, an enamel shade can be recognized only along interdental tooth surfaces, (e) A backscattered electron micrograph from an impacted maxillary third molar reveals that the mineral density of the inner 2/3 to 3/4 of the enamel (E) is considerably reduced (arrows), (g) The detail marked by the rectangle in (e) shows that the mineral deficiency is not uniform, but particularly prominent along the borders of the enamel prisms (arrows). This may account for the disproportionate reduction in biomechanical properties, (b) Frontal view of the permanent incisors of a male patient. In contrast to hypomaturated (a), hypocalcified enamel exhibits a yellow-brown discoloration and is so soft that it does not break, but is rapidly lost as a result of masticatory function. Note the primary molars which seem markedly less affected, (d) The panoramic radiograph from the same patient does not allow discriminating enamel from dentin, (f) As shown by a light micrograph from a ground section of a maxillary primary canine, the hypocalcified enamel (E) is stained as intensely as dentin (D), because it contains large amounts of organic matrix, (h) In contrast, normal enamel (E) is not stained at all, although coloration of the dentin (D) is comparable to that shown in (f). P, pulp. Original magnifications (e) x25, (f) x50, (g) xllOO, (h) xl2.5.
|
Fig. 5. Hypomaturated (a, c, e, g) and hypocalcified (b, d, f) forms of amelogenesis imperfecta, (a) Lateral view of the posterior permanent teeth of a female patient showing extensive enamel chipping, because hypomaturated enamel exhibits reduced resistance to mechanical loading, but is hard enough to break, (c) In the panoramic radiograph from the same patient, an enamel shade can be recognized only along interdental tooth surfaces, (e) A backscattered electron micrograph from an impacted maxillary third molar reveals that the mineral density of the inner 2/3 to 3/4 of the enamel (E) is considerably reduced (arrows), (g) The detail marked by the rectangle in (e) shows that the mineral deficiency is not uniform, but particularly prominent along the borders of the enamel prisms (arrows). This may account for the disproportionate reduction in biomechanical properties, (b) Frontal view of the permanent incisors of a male patient. In contrast to hypomaturated (a), hypocalcified enamel exhibits a yellow-brown discoloration and is so soft that it does not break, but is rapidly lost as a result of masticatory function. Note the primary molars which seem markedly less affected, (d) The panoramic radiograph from the same patient does not allow discriminating enamel from dentin, (f) As shown by a light micrograph from a ground section of a maxillary primary canine, the hypocalcified enamel (E) is stained as intensely as dentin (D), because it contains large amounts of organic matrix, (h) In contrast, normal enamel (E) is not stained at all, although coloration of the dentin (D) is comparable to that shown in (f). P, pulp. Original magnifications (e) x25, (f) x50, (g) xllOO, (h) xl2.5.
во время стадии созревания. Ген для наиболее обильного эмалевого матричного белка,
amelogenin, локализуется на X- и Y-хромосомах, но у особей мужского пола 90% белка транскрибируется с
AMELX, копии на X хромосоме (96, 97). Следовательно, мутации в
AMELX будут вызывать X-сцепленную AI, которая дает разнообразные фенотипы в зависимости от пола и места мутации (96). В частности, гипопластические формы AI отличаются у разных полов. В то время как у самцов присутствует smooth hypoplastic AI, фенотип у самок характеризуется вертикальными бороздами, которые обусловлены инактивацией Х хромосомы (Fig. 4b) (98). Мутации в сигнальном пептиде AMELX приводят к недостаточности секреции белка, а также к дефектам, которые укорачивают критический С-конец amelogenin, вызывая smooth hypoplastic AI. Напротив, мутации в N-терминальной области AMELX, которые удаляют или изменяют сайт proteinase расщепления, дают недозрелого типа AI (97, 99).
 Figure 6. Axon viability depends on coordinated endocytic trafficking and signaling. Nerve growth factor (NGF) binds to the TrkA receptor tyrosine kinase and stimulates phosphorylation and internalization. Endocytosed TrkA is transported from Rab5-positive early endosomes to Rab7-positive late endosomes. In peripheral neurons long distance transport of late endosomes to the cell body is critical for growth factor degradation and proper nuclear signaling to maintain cell viability and differentiation. Transport occurs on microtubules through Rab7, the Rab interacting lysosomal protein (RILP) effector and dynactin minus-end directed motor complex. Return transport to the synapse is mediated by Rab7 in conjunction with a plus-end directed kinesin motor, likely KIF3a. Inset illustrates endosomal membrane protein complexes involved in Rab conversion that allows transfer of cargo along the degradative pathway. Several conserved multimeric protein complexes first identified in yeast are thought to aid in Rab conversion, directed transport and fusion (RETROMER endosome to Golgi; CORVET early to late endosome; HOPS late endosome to lysosome).
Figure 6. Axon viability depends on coordinated endocytic trafficking and signaling. Nerve growth factor (NGF) binds to the TrkA receptor tyrosine kinase and stimulates phosphorylation and internalization. Endocytosed TrkA is transported from Rab5-positive early endosomes to Rab7-positive late endosomes. In peripheral neurons long distance transport of late endosomes to the cell body is critical for growth factor degradation and proper nuclear signaling to maintain cell viability and differentiation. Transport occurs on microtubules through Rab7, the Rab interacting lysosomal protein (RILP) effector and dynactin minus-end directed motor complex. Return transport to the synapse is mediated by Rab7 in conjunction with a plus-end directed kinesin motor, likely KIF3a. Inset illustrates endosomal membrane protein complexes involved in Rab conversion that allows transfer of cargo along the degradative pathway. Several conserved multimeric protein complexes first identified in yeast are thought to aid in Rab conversion, directed transport and fusion (RETROMER endosome to Golgi; CORVET early to late endosome; HOPS late endosome to lysosome).
Поскольку пока не обнаружено дефектов, ответственных за AI, в гене, кодирующем AMBN, несколько амелобластных мутаций идентифицировано в гене enamelin (ENAM), расположенном на хромосоме 4q21 (97, 99-102). Они проводят к аутосомно доминантной или рецессивной гладкой или грубой гипопластической AI. В то время как гладкий гипопластический фенотип напоминает тот. что наблюдается при мутациях AMELX, грубая гипопластическая AI, вызываемая дефектами в ENAM, обнаруживает своеобразный фенотип, характеризующийся горизонтальными углублениями (103). Было предположено, что дефекты эмали, обусловленные мутациями ENAM, являются зависимыми от дозы, гладкий и грубый гипопластические фенотипы сегрегируют как рецессивный и доминантный признак, соотв. (100).
Др. группа мутаций, вызывающая AI затрагивает гены enamelysin (MMP20) и kallikrein 4 (KLK4), каждая из которых специфична для зубов и важна для собственно созревания эмали (96, 99, 104, 105). Фактически, все идентифицированные генетические дефекты приводят к потере функции энзимов и к аутосомно рецессивной AI (пигментированного) недозрелого типа. Довольно неожиданно некоторые мутации, вызывающие очень сходные фенотипы AI были недавно идентифицированы в WDR72, в гене для белка. содержащего WD повторы 72 (106, 107). Он в самом деле экспрессируется на стадии созревания амелобластов (107), но пока неизвестно его участие в образовании эмали.
Мутации, отвечающие за аутосомно доминантную гипокальцифицированную AI, которая является третьей основной формой AI и наиболее широко распространена в Сев. Америке, были идентифицированы лишь недавно. Kim et al. (108) описали две nonsense мутации в FAM83H (family with sequence similarities 83 member H), которые в точности сегрегируют с болезнью. Идентификация дополнительных дефектов в том же самом гене (109-112) позволила выявить генотип-фенотипические корреляции (113). Все мутации, большинство из которых nonsense мутации, возникают в последнем экзоне и существенно укорачивают предполагаемый белок. Было предположено, что короткий белок вызывает генерализованную гипокальцифицированную AI, тогда как менее тяжело укороченный белок ведет к бросающемуся в глаза ослабленному фенотипу, характеризующемуся гипокальцифицированной эмалью, ограниченной шеечной частью коронки (113). Поскольку ген FAM83H экспрессируется не только в зубах (108), то возникает вопрос, почему мутации, вызывающие AI, не оказывают каких-либо очевидных последствий на др. ткани. Предполагается, что минерализация эмали критически зависит от высоких уровней белка и что укороченный белок может оказывать доминантный негативный эффект (109, 113).
Итак, разные фенотипические формы AI являются генетически гетерогенными. Мутации и в AMELX и в ENAM могут вызывать или гладкую или грубую гипопластическую AI, а недозрелый тип может вызываться генетическими дефектами в
AMELX, MMP20, KLK4 и WDR72. Лишь мутации в FAM83H, по-видимому,
последовательно приводят к гипокальцифицированной AI, хотя и с некоторой изменчивостью в экспрессивности. Однако в целом менее половины случаев AI может быть объяснено дефектами известных генов кандидатов (114, 115).
Conclusion
Over the last years, a big effort has been made to understand the molecular and cellular mechanisms controlling tooth development and pathology. Much information on the genes that are important for human tooth formation has been revealed using the mouse model. However, very little is known on the generation of well-known human dental pathologies that are the consequence of aberrant cell differentiation and subsequent dental matrix formation. The complex genetic interactions leading to these human dental malformations can be only studied in detail in transgenic mice. Elucidating when and how signaling molecules and transcription factors dictate tooth initiation, morphology and mineralization will open new horizons to the dental discipline and will create new challenges. Novel genetic knowledge together with tissue engineering and stem cell approaches will probably instruct development of novel therapies in dentistry (116, 117).
Сайт создан в системе
uCoz
 |
Fig. 1. Stages of embryonic human tooth development. Dental epithelium and its derivatives (enamel) are in red color, dental mesenchyme and its derivatives (dentin) in blue. The most significant signaling molecules (in bold capitals) and transcription factors (in italics) that are involved in the various stages of tooth development are shown, epithelial signals and transcription factors in red. mesenchymal signals and transcription factors in blue. Mutations in humans affecting tooth development are presented with asterisks.
|
Fig. 1. Stages of embryonic human tooth development. Dental epithelium and its derivatives (enamel) are in red color, dental mesenchyme and its derivatives (dentin) in blue. The most significant signaling molecules (in bold capitals) and transcription factors (in italics) that are involved in the various stages of tooth development are shown, epithelial signals and transcription factors in red. mesenchymal signals and transcription factors in blue. Mutations in humans affecting tooth development are presented with asterisks.  Fig. 2. Expression of transcription factors in the ectomesenchyme and epithelium of the nasal (np), maxillary (mx) and mandibular (md) processes during embryogenesis. (a) Neural crest-derived cells that migrate into the first branchial arch of an embryo are under the influence of signaling molecules and transcription factors (indicated by a variety of colors). Disposal of the various teeth on the dental axis is time-dependent, and combinations of different signaling molecules and transcription factors will contribute to a variety of tooth shapes/classes (incisors, canines, premolars and molars). Defects in the number of neural crest cells, signals (e.g. SHH) or transcription factors (e.g. MSX1) are responsible for misshaped teeth and/or tooth agenesis, (b) Expression of Barxl in the first branchial arch mesenchyme of a mouse embryo, (c) Expression of Tbxl in oral epithelium of a mouse embryo, oc, oral cavity.
Fig. 2. Expression of transcription factors in the ectomesenchyme and epithelium of the nasal (np), maxillary (mx) and mandibular (md) processes during embryogenesis. (a) Neural crest-derived cells that migrate into the first branchial arch of an embryo are under the influence of signaling molecules and transcription factors (indicated by a variety of colors). Disposal of the various teeth on the dental axis is time-dependent, and combinations of different signaling molecules and transcription factors will contribute to a variety of tooth shapes/classes (incisors, canines, premolars and molars). Defects in the number of neural crest cells, signals (e.g. SHH) or transcription factors (e.g. MSX1) are responsible for misshaped teeth and/or tooth agenesis, (b) Expression of Barxl in the first branchial arch mesenchyme of a mouse embryo, (c) Expression of Tbxl in oral epithelium of a mouse embryo, oc, oral cavity.  |
Fig. 3. Panoramic radiographs illustrating types of agenesis of permanent teeth (asterisks), (a) Single agenesis of the lower left second premolar, the most frequently missing permanent tooth apart from the third molars, (b) Oligodontia as it is known to occur due to mutations in MSX1: All the eight premolars, two of four canines and seven of eight incisors, but no molars are missing, (c) Oligodontia as it could occur due to a defect in PAX9: All upper molars, but no other permanent teeth are missing (for a reliable assessment of third molar tooth buds, the patient was too young).
|
Fig. 3. Panoramic radiographs illustrating types of agenesis of permanent teeth (asterisks), (a) Single agenesis of the lower left second premolar, the most frequently missing permanent tooth apart from the third molars, (b) Oligodontia as it is known to occur due to mutations in MSX1: All the eight premolars, two of four canines and seven of eight incisors, but no molars are missing, (c) Oligodontia as it could occur due to a defect in PAX9: All upper molars, but no other permanent teeth are missing (for a reliable assessment of third molar tooth buds, the patient was too young).