Посещений:  ПОДЖЕЛУДОЧНАЯ ЖЕЛЕЗА: β-КЛЕТКИ
ПОДЖЕЛУДОЧНАЯ ЖЕЛЕЗА: β-КЛЕТКИ
Роль Nkx6.1
|
|
Nkx6.1 Is Essential for Maintaining
the Functional State of Pancreatic Beta Cells BrandonL. Taylor, Fen-Fen Liu, andMaikeSander  Cell Reports (2013), http://dx.doi.org/10.1016/j.celrep.2013.08.010 |
Recently, loss of beta-cell-specific traits has been
proposed as anearly cause of beta cell failure in diabetes. However, the molecular mechanisms that underlie the loss of beta cell features remain unclear. Here, we identify an Nkx6.1-controlled gene regulatory network as essential for maintaining the functional and molecular traits of mature beta cells.Conditional Nkx6.1inactivation in adult mice caused rapid-onset diabetes and hypoinsulinemia. Genomewide analysis of Nkx6.1-regulated genes and functional assays further revealed a critical role forNkx6.1 in the control of insulin biosynthesis, insulinsecretion, and beta cell proliferation. Over time,Nkx6.1-deficient beta cells acquired molecular characteristics of delta cells, revealing a molecular link between impaired beta cell functional properties and loss of cell identity. Given that Nkx6.1 levelsare reduced in human type 2 diabetic beta cells,our study lends support to the concept that loss ofbeta cell features could contribute to the pathogenesis of diabetes.

|
Типа 2 diabetes mellitus (T2D) характеризуется снижением чувствительности к инсулину Хотя оба эти фактора играют роль, генетические исследования подтвердили, что способность beta клеток отвечать на метаболические стрессоры является доминирующим фактором в детерминации предрасположенности к T2D (Muoio and Newgard, 2008). При T2D, beta клетки обнаруживают нарушенную способность компенсировать повышенные потребности в инсулине (Cerasi and Luft, 1967), дефект, который был приписан как неадекватной клеточной способности секретировать инсулин (Hosker et al., 1989) , так и гибели beta клеток (Butler
etal.,2003). Среди наиболее ранних дефектов, наблюдаемых у T2D пациентов это снижение способности beta клеток секретировать инсулин в ответ на повышенные уровни глюкозы в крови (Hosker et al., 1989). Это нарушение в секреции инсулина, стимулируемой глюкозой, было приписано дефектам ощущения глюкозы (Frogueletal.,1992), дисфункцией митохондрий
(Supale et al., 2012) и оксидативным стрессам (Robertson, 2004). Т.о. огромное большинство доказательств указывает на то, что дефекты во многих клеточных процессах могут нарушить функцию beta клеток и могут служить фактором возникновения T2D. Более того, гипергликемия, как было показано, нарушает экспрессию генов, которые важны для качественных характеристик beta клеток (Jonas et al., 1999). Недавно, Talchai et al. (2012) описали потерю свойств beta клеток, характеризующуюся снижением продукции инсулина, приобретением характеристик, подобных таковым у предшественников и изменение судьбы в др. типы эндокринных клеток у мышей, моделирующих T2D, это указывает на то, что потеря дифференцированного состояния beta клетками также вносит вклад в недостаточность beta клеток при T2D. Однако сегодня неизвестно, потеря ли функциональных свойств beta клеток (а именно, регулируемую секрецию инсулина) или потеря качественных особенностей beta клеток связаны с прогрессированием T2D. Одновременная потеря функции beta клеток и качественных особенностей д. объяснить пониженную экспрессию центрального транскрипционного регулятора, контролирующего гены, участвующие в обоих процессах.
Несколько линий доказательств указывают на то, что концентрирующийся в beta клетках транскрипционный фактор Nkx6.1 д. играть роль в T2D. Во-первых, исследования геномных ассоциаций подтверждают, что варианты Nkx6.1 ассоциируют с T2D (Yokoi et al., 2006). Во-вторых, пониженная экспрессия Nkx6.1, как было установлено, сопровождает возникновение T2D у грызунов и человека (Guo et al., 2013; Talchai et al., 2012). В-третьих, исследования in vitro линий beta клеток и изолированных островков подтверждают возможную роль Nkx6.1 в регуляции стимулируемой глюкозой секреции инсулина, а также пролиферации beta клеток (Schisler et al., 2005, 2008). Кроме того, мы недавно показали, что Nkx6.1 необходим и достаточен для обеспечения характеристик beta клеток в дифференцирующихся эндокринных предшественниках у эмбрионов (Schaffer et al., 2013), это открывает возможность, что Nkx6.1 может также помогать поддерживать дифференцированное состояние beta клеток взрослых. Итак, эти находки подтверждают, что Nkx6.1 может быть регулятором функции и качественных особенностей beta клеток у взрослых животных.
Чтобы исследовать роль Nkx6.1 в зрелых beta клетках, мы устраняли специфически Nkx6.1 в beta клетках взрослых мышей и установили гены мишени для Nkx6.1 в beta клетках с помощью комбинации профилирования экспрессии генов и иммунопреципитации хроматина с массивным параллельным секвенированием (ChIP-seq). Мы установили, что потеря Nkx6.1 вызывает быстро начинающийся диабет, обусловленный дефектами биосинтеза и секреции инсулина. Наблюдаемые потеря продукции инсулина и функциональных свойств beta клеток позднее сопровождались эктопической активацией генов delta клеток в beta клетках. Т.о., в результате нарушения функции beta клеток и дестабилизации качественных особенностей beta клеток, пониженные уровни Nkx6.1, как это наблюдается при T2D, д. вносить вклад в патогенезT2D.
DISCUSSION
Хорошоо известно, что дисфункция beta клеток, особенно неспособность beta клеток собственно секретировать инсулин в ответ на высокие уровни глюкозы в крови, является наиболее ранним клиническим проявлением в процессе развития T2D (Ferrannini, 2010). Способностью, ощущать уровни глюкозы и увязывать эту информацию с секреторным инсулиновым ответом, наделены beta клетки благодаря специализированным транспортерам и энзимам. Хотя механизмы, которые лежат в основе обуславливаемой глюкозой секреции инсулина, довольно хорошо известны, но остается неясным, какие регуляторы транскрипции инициируют и поддерживают экспрессию генов, которые позволяют beta клеткам осуществлять свою чрезвычайно специализированную функцию. В данном исследовании мы показали, что транскрипционный фактор Nkx6.1 является главным регулятором генов, которые предопределяют функциональное состояние beta клеток, эта роль согласуется с его исключительной экспрессией в beta клетках поджелудочной железы взрослых.
Мы показали, что кондиционная инактивация Nkx6.1 в beta клетках взрослых мышей ведет к сахарному диабету в течение дней после удаления Nkx6.1. Потеря активности Nkx6.1 оказывает непосредственный и драматический эффект на экспрессию генов, которые придают beta клеткам их уникальную способность синтезировать и высвобождать инсулин регулируемым способом. Мы установили, что гены, участвующие в биосинтезе инсулина (Slc30a8 и Ero1lb), импорте глюкозы (Glut2) и метаболизме глюкозы (Pcx) являются непосредственными транскрипционными генами мишенями для Nkx6.1. Кроме того, устранение Nkx6.1 косвенно влияет на экспрессию многочисленных генов, которые важны для функции beta клеток м что интересно, также для пролиферации beta клеток (Figure 7). Находка, что уровни островкового Ccnd2 и пролиферация beta клеток снижены у кондиционных мутантных Nkx6.1 мышей, оказалось до некоторой степени неожиданностью, поскольку некоторые исследования показали, что гипергликемия оказывает стимулирующий эффект на экспрессию в beta клетках Ccnd2 и их самообновление (Alonso et al., 2007; Bonner-Weir et al., 1989; Salpeter et al., 2011). Мы установили, что снижение функциональности beta клеточного митогена инсулина (Paris et al., 2003) не вносит очевидного вклада в снижение пролиферативной способности beta клеток после устранения Nkx6.1. Вместо этого наши результаты показали, что пролиферативная способность дефицитных по Nkx6.1 beta клеток ограничивается за счет снижения внутриклеточной функции глюкозы из-за потери экспрессии Glut2 (Figure 7). Эти находки демонстрируют внутреннюю связь между способностью beta клеток импортировать глюкозу и их пролиферативной способностью, это ведет к дальнейшему подтверждению недавней концепции, что метаболизм глюкозы играет критическую роль в регуляции пролиферации beta клеток (Porat et al., 2011; Salpeter et al., 2010, 2011). В комбинации с находкой, что уровни Nkx6.1 понижены у T2D моделей метаболических стрессов (Talchai et al., 2012), наша работа подтверждает, что Nkx6.1 действует как метаболический сенсор, который модулирует как секрецию инсулина, так и пролиферативную способность в ответ на метаболические стрессы. С помощью предупреждения пролиферации beta клеток, которые теряют чувствительность к глюкозе, клеточно автономная связь между импортом глюкозы в beta клетки и их пролиферацией может представлять наследуемый механизм селекции здоровых beta клеток.
Наше исследование также примирило ранее описанные противоречивые находки о роли Nkx6.1 в пролиферации beta клеток. Мы недавно установили, что избыточная экспрессия трансгена Nkx6.1 в beta клетках взрослых мышей in vivo не оказывает позитивного эффекта на пролиферацию beta клеток или массу beta клеток (Schaffer et al., 2011). Напротив, вирусом обусловленная экспрессия Nkx6.1 в культивируемых островках вызывала пролиферативную активность (Schisler et al., 2008). Возможным объяснением этому противоречию является то, что культура in vitro островков нарушает уровни экспрессии Nkx6.1. Известно, что как только они удаляются из своих ниш и помещаются в культуру, то beta клетки быстро теряют экспрессию Glut2 и прекращают пролиферацию (Weinberg et al., 2007), указывая тем самым, что экспрессия вышестоящих регуляторов Glut2, включая Nkx6.1, также нарушена. Т.о., наблюдаемый пролиферативный эффект Nkx6.1 in vitro может быть объяснен экспрессией вирусами Nkx6.1 , восстанавливающей пониженные уровни Nkx6.1 и, в свою очередь, также уровни Glut2 и Ccnd2 в культивируемых островках. Напротив, повышенные уровни Nkx6.1 выше нормы в здоровых beta клетках in vivo, по-видимому, не оказывают дальнейшего стимулирующего эффекта на импорт глюкозы и пролиферацию beta клеток.
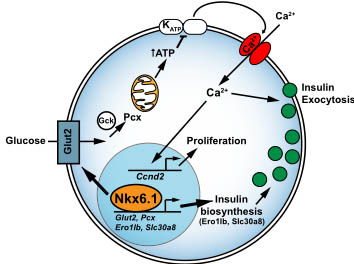 Figure 7. Nkx6.1 Function in Adult Beta Cells Figure 7. Nkx6.1 Function in Adult Beta Cells
Nkx6.1 directly regulates transcription of genes encoding proteins involved in glucose uptake and metabolism (Glut2 and Pcx) and insulin biosynthesis (Ero1lb and Slc30a8). Reduced expression of these Nkx6.1 target genes affects beta cell function in three ways: First, decreased glucose uptake and metabolism diminishes ATP production, leading to impaired insulin secretion via the stimulus-secretion coupling pathway. Second, by enabling glucose uptake through Glut2 regulation, Nkx6.1 indirectly controls beta cell proliferative capacity. In the absence of Nkx6.1, expression of Ccnd2, which encodes the critical beta cell mitogen Cyclin D2, is reduced and beta cell proliferation is decreased. Reconstituting Glut2 expression in Nkx6.1-deficient beta cells restores Ccnd2 levels and beta cell proliferation. Third, insulin biosynthesis is severely impaired, leading to reduced production of mature insulin and an overabundance of immature secretory vesicles. Gck, glucokinase; Glut2, glucose transporter 2; KATP, ATP-sensitive potassium channel; Pcx, pyruvate carboxylase.
После делеции Nkx6.1, мы наблюдали чрезвычайно быстрое снижение содержания инсулина, при этом потеря инсулина не сопровождалась гибелью beta клеток, демонстрируя сильное сходство между дефицитными по Nkx6.1 beta клетками и ''пустыми'' beta клетками, наблюдаемыми у мышей, моделирующих T2D (Talchai et al., 2012). Более того, как сообщалось, в условиях метаболического стресса (Talchai et al., 2012), мы установили, что пониженная экспрессия Nkx6.1 сопровождается также дерепрессией маркера клеток эндокринных предшественников Ngn3. Базируясь на наблюдении, что beta клетки подвергаются изменению судьбы в не-beta эндокринные типы клеток у T2D моделей (Talchai et al., 2012), потеря признаков beta клеток и избыточная экспрессия Ngn3, как полагают, делают beta клетки пластичными и и более склонными к изменению своих качественных особенностей. В соответствии с этим мнением, мы наблюдали, что субнабор Nkx6.1-дефицитных beta клеток эктопически экспрессирует somatostatin спустя 8 недель после делеции Nkx6.1. Ранее мы показали при отслеживании клонов, что делеция Nkx6.1 в эмбриональных beta клетках ведет к быстрому переключению судеб с beta-на-delta клетки (Schaffer et al., 2013). Однако превращение beta клеток в др. не-beta эндокринные типы клеток не наблюдается после инактивации Nkx6.1 в эмбриональных beta клетках. Сходным образом, после делеции Nkx6.1 в beta клетках взрослых, мы наблюдали совместную экспрессию инсулина исключительно со somatostatin, но не с др. панкреатическими гормонами. Т.о., потеря Nkx6.1 ведет к избирательной дерепрессии генов delta клеток как в эмбриональных, так и во взрослых beta клетках. Однако превращение судьбы более полное и происходит более быстро, когда Nkx6.1 инактивирован в незрелых beta клетках. Следовательно, последовательная потеря признаков beta клеток предшествует принятию альтернативной судьбы эндокринных клеток, наблюдаемому после инактивации Nkx6.1 у взрослых, в точности отображает постепенную потерю функциональной массы beta клеток, наблюдаемую у моделей T2D (Talchai et al., 2012).
Наше исследование подтвердило концепцию, что транскрипционные факторы, такие как Nkx6.1 и FoxO1 (Talchai et al., 2012), являются критическими для поддержания beta клеток в их дифференцированном состоянии. Ключевым вопросом, который нуждается в дальнейшем исследовании, является может ли дестабилизированное состояние beta клеток, наблюдаемое у человека, вносить возможный вклад в патогенез T2D. Наблюдение, что экспрессия NKX6.1 снижается в beta клеток у пациентов с T2D (Guo et al., 2013) подтверждает, что находки у грызунов могут, в самом деле, иметь значение для болезней у человека. Необходимы дальнейшие исследования для выяснения, какие аспекты фенотипа у грызунов обнаруживаются и у людей и как потеря признаков beta клеток связана с прогрессированием болезни. Такое знание поможет идентифицировать период терапевтического вмешательства, во время которого функциональное состояние beta клеток может быть восстановлено прежде, чем beta клетки превратятся в др. типы эндокринных клеток.
|