Посещений:  КЛЕТОЧНОЕ СТАРЕНИЕ
КЛЕТОЧНОЕ СТАРЕНИЕ
Роль в развитии
|
|
A New Development in Senescence Ana Banito and Scott W. Lowe  Cell 755, P. 977-978. November 21, 2013 |
|
|
Клеточное старение участвует в нескольких патологических реакциях у взрослых с важными последствиями на супрессию опухолей, заживление ран и старение. Два исследования Muhoz-Espin et al. и Storer et al. показали, что старение вносит вклад в эмбриональное развитие, подтверждая его изначальную роль в нормальной физиологии.
Плодотворное исследование Hayflick and Moorhead продемонстрировало, что нормальные клетки могут только делиться определенное количество раз, прежде чем они достигнут состояния репликативного клеточного старения. Хотя сегодня мы верим, что старение играет широкую роль в различных стрессовых реакциях у взрослых, но в данном номере ж., Munoz-Espin et al. (2013) и Storer et al. (2013) сообщают о неожиданных результатах, что старение происходит при физиологических условиях во время эмбрионального развития млекопитающих.
Клетки подвергаются репликативному старению, подавляя гены клеточного цикла и определенные компоненты внеклеточного матрикса, усиливая в то же время активность генов, кодирующих ингибиторы клеточного цикла, энзимов, деградирующих матрикс, в частности цитокинов и факторов иммунного надзора. Клеточные стрессы, такие как раскрытие (uncapping) теломер или активация онкогенов, могут запускать программы стабильного ареста клеточного цикла со сходными свойствами, хотя существуют споры о существовании разных "типов" старения (Shay and Roninson, 2004). Наиболее широко используемый маркер старения это ассоциированная со старением активность senescence-associated p-galactosidase (SApG), которая, скорее всего, отражает появление повышенной аутофагии в стареющих клетках (Young et al., 2009). Др. каноническими маркерами старения являются p53, p21, p16 и reduced RB фосфорилирование, которые коллективно обусловливают дополнительные фенотипические проявления, ассоциированного со старением ареста клеточного цикла. Затронутые клетки часто накапливают гетерохроматиновые фокусы, которые могут стабилизировать состояние старости и они обнаруживают измененные профили секреции, которые модулируют иммунную функцию и/или укрепляют арест клеточного цикла (Kuilman et al., 2010). Концептуальная проблема заключается в том, что ни один из этих маркеров не являются уникальным для стареющих клеток и нет единственного маркера, достаточного для "диагностики" состояния старости. Как результат старение определяется с помощью коллекции маркеров, которые не являются убедительными.
Старение в основном рассматривается как программа стрессовой реакции. Пока что намеки, что старение может играть некую физиологическую роль, исходили из исследований вовлечения старения в ограничение определенных реакций заживления ран (Jun and Lau, 2010; Krizhanovsky et al., 2008). Хотя активность SApG была описана в регрессирующих мезонефросах у птиц (Nacher et al., 2006), его значение у эмбрионов млекопитающих оставалось неизвестным. Новые сообщения, предполагают, что старение происходит в ходе всего развития мыши. Munoz-Espin et al. сфокусировались на внутреннем ухе и регрессии мезонефрических канальцев, тогда как Storer et al. сконцентрировались на apical ectodermal ridge (AER) во время формирования конечности. Оба исследования предполагали, что "онтогенетическое старение" обладает некоторыми общими, но не всеми, регуляторными путями, наблюдаемыми у взрослых (Figure 1).
Оба состояния старения обладают одинаково активностью SApG и ассоциированными со старением гетерохроматиновыми маркерами (HP1y и H3K9me3) и оба обнаруживают снижение окрашивания Ki67 (маркера пролиферации) из-за ареста в G1. Однако онтогенетическое старение, по-видимому, не участвует в активации p16 или p19ARF и не запускается с помощью p53 или повреждения ДНК. Вместо этого онтогенетическое старение обеспечивается с помощью p21 независимым от p53 способом, но контролируется вместо этого с помощью TGFβ/SMAD- и PI3K/FOXO-сигнальных путей. Хотя стареющие клетки у эмбрионов и взрослых, каждый секретирует факторы, которые заставляют иммунную систему элиминировать клетки и ремоделировать ткани, секретируемые цитокины и ростовые факторы не все те же самые (Figure 1).
Возникает вопрос, действительно ли эти феномены в самом деле представлены разными типами старения или отражают фундаментально различные процессы. В соответствии с представленными выше наблюдениями, p53 или Ink4a/Arf нокаутные мыши не представляют собой альтерации паттернов активности SApG во время развития и не проявляют аномалий в тканях, в которых наблюдается старение. Однако, p21 нулевые эмбрионы обнаруживают меньше SApG-позитивных клеток по сравнению с контролем и обнаруживают четкие онтогенетические аномалии в ассоциированных тканях. Всё же многие из этих эмбриональных дефектов исправляются у новорожденных. Имеются, по крайней мере, две правдоподобные причины, почему фенотип p21 нулевых мышей может не позволять считывание потенциально важных программ. Во-первых, возможно, что p21 делеция недостаточна, чтобы отвергнуть старение или может только задерживать его индукцию. Во-вторых, эмбрион может компенсировать потерю p21, задействуя альтернативные ткань-моделирующие программы.
Вероятность, что компенсаторные механизмы могут маскировать ключевую роль определенных программ в развитии не без прецендента. Существуют неотразимые доказательства важности апоптоза в эмбриональном развитии; хотя, разрушение прирожденных апоптических программ у эмбрионов дает лишь умеренные фенотипы. В качестве примера, апоптоз рассматривается как основной механизм клеточной гибели в развивающихся конечностях, но инактивация проапоптических генов у мышей лишь частично предупреждает удаление межпальцевых тканей (Fuchs and Steller, 2011). Очевидно, существует компенсаторная программа, чтобы инструктировать морфогенез
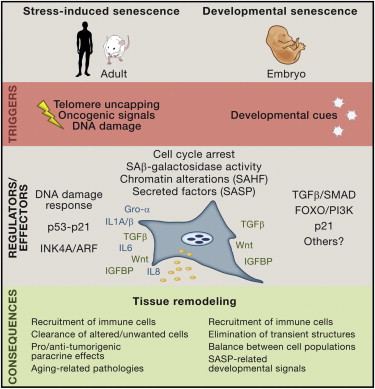 Figure 1. Main Features of Senescence in the Adult and in the Embryo
Figure 1. Main Features of Senescence in the Adult and in the Embryo
Whereas the first is induced by stress, such as telomere uncapping, oncogenic signals, or DNA damage, the second is induced by still undetermined developmental cues. Both programs share a set of features such as senescence-associated li-galactosidase activity (SA(iG), senescence-associated heterochromatic foci (SAHF), and some of the members of the senescence-associated secretory phenotype (SASP) such as TGFfi, Wnt, and IGFBP family ligands. Nevertheless, differences exist. Developmental senescence does not depend on the activation of DNA damage response, p53-21, or p16 tumor suppressor pathways and does not present some of the SASP-related factors such as IL8 (Cxcll, 2, and 5 homologs in mice) and IL6. Although additional regulators may exist, senescence in the embryo is mainly mediated by p21 and regulated by the TGF|VSMAD- and FOXO/PI3K-signaling pathways. Tissue remodeling is a main consequence of both programs. By recruiting the immune system, senescence mediates the elimination of unwanted/transient cells or structures. Developmental senescence may additionally dictate the balance between cell populations or instruct developmental processes.
взрослых, развившаяся из программы ремоделирования примордиальных тканей, которые имеются у эмбрионов. В обеих программах клетки арестовываются во время клеточного цикла, частично обладают общим набором функциональных маркеров, играют активную роль в модификации тканевого микроокружения и в конечном итоге распознаются и очищаются с помощью иммунной системы (Figure 1). Эти свойства могут быть адаптированы как часть возникающей у взрослых программы стрессовой реакции, которая включает в себя дополнительные механизмы супрессии опухолей, такие как те, что базируются на p53 и p16, чтобы элиминировать поврежденные клетки и это может , в свою очередь, вносить вклад в старение организма. Эти исследования представляют др. характерное свойство в области старения, когда ткань ремоделируется в отсутствие апоптоза. Напротив, неспособность старения в p21 нулевых мезонефрических канальцах сопровождается задержкой активации апоптоза и макрофагами обеспечиваемой очистки погибших клеток. Было бы интересно установить, может ли старение компенсироваться недостаточностью апоптоза во время развития.
Итак, какова потенциальная роль старения в эмбриональном развитии? То, что старение и инфильтрация макрофагов предшествуют инволюции мезонефросов, подтверждает, что одной из ролей старения является ремоделирование эмбриональных почек. Старение может также обладать инструктивной функцией. В самом деле, Storer et al. нашли, что сигнатура экспрессии в AER частично перекрывается с таковой, индуцируемого онкогенами старения, указывая, что секретируемые компоненты из стареющих клеток влияют на формирование паттерна и пролиферацию соседней мезенхимы.
Наконец, путем удержания пролиферации специфических клеток внутри развивающихся тканей, старение может диктовать сбалансированный рост и взаимодействие между разными популяциями клеток. Напр., этот феномен может иметь место в эндолимфатическом мешке, который не элиминируется во время развития, а вместо этого подвергается процессу дифференциального клеточного пролиферативного ареста, который изменяет относительные количества разных клеточных популяций.
Вообще-то наиболее важное ответвление новых работ связано с их значением для эволюционного происхождения программ старения. Большинство исследований, кстати, было сфокусировано на старении как процессе супрессии опухолей и дебатах, как эволюция отбирала программы, которые бы предупреждали нарушения, которые обычно появляются после репродуктивного возраста (Campisi, 2003). Новые работы открывают возможность, что старение также откроет новый круг относящихся к делу вопросов. Каковы онтогенетические сигналы, которые запускают старение у эмбрионов и до какой степени этот процесс отражает программу старения, индуцируемую стрессами, изученную подробно? Каковы существенные характеристики, которые предопределяют состояние клеточного старения?
REFERENCES
Campisi, J. (2003). Nat. Rev. Cancer 3, 339-349.
Fuchs, Y„ and Steller, H. (2011). Cell 147, 742-758.
Jun, J.I., and Lau, L.F. (2010). Nat. Cell Biol. 12, 676-685.
Krizhanovsky, V., Yon, М., Dickins, R.A., Hearn, S., Simon, J., Miething, C., Yee, H., Zender, L., and Lowe, S.W. (2008). Cell 134, 657-667. Kuilman, Т., Michaloglou, C., Mooi, W.J., and Peeper, D.S. (2010). Genes Dev. 24, 2463-2479. Mufioz-Espin, D., Canamero, М., Maraver, A., Gomez-Lopez, G., Contreras, J., Murillo-Cuesta, Rodriguez-Baeza, A., Varela-Nieto, I., Rubert, J., Collado, М., et al. (2013). Cell 155, this issue, 1104-1118.
Nacher, V., Carretero, A., Navarro, М., Armengol, C., Llombart, C., Rodriguez, A., Herrero-Fresneda, I., Ayuso, E., and Ruberte, J. (2006). J. Vase. Res. 43, 581-586.
Shay, J.W., and Roninson, I.B. (2004). Oncogene 23, 2919-2933.
Storer, М., Mas, A., Robert-Moreno, A., Pecoraro, М., Ortells, M.C., Di Giacomo, V., Yosef, R., Pilpel, N.. Krizhanovsky, V., Sharpe, J., et al. (2013). Cell 155, this issue, 1119-1130.
Young, A.R., Narita, М., Ferreira, М., Kirschner, K., Sadaie, М., Darot, J.F., Tavare, S., Arakawa, S., Shimizu, S., Watt, F.M., and Narita, M. (2009). Genes Dev. 23, 798-803.
|