Это цитата из сев. европейского фольклора, является ранним отражением этой общераспространенной болезни, называемой сегодня кистозным фиброзом. Это заболевание, приводящее к гибели детей в раннем возрасте, часто выявляется по избыточному выделению солей с потом. Соленое чело одно из наиболее злокачественных проявлений. Наследуемые генетические аномалии могут также нарушать легкие и вызывать серьезные дефекты в поджелудочной железе, тонком кишечнике и печени. Достижения в терапии на протяжении последних нескольких десятилетий были обогащены наблюдениями за больными детьми, оказавшимися способными более, чем в половине случаев доживать до 20 и более лет.
Но не существует адекватного лечения, которое могло бы исправить биохимический дефект в корне, и ничто не может отодвинуть спектр детской смертности.
Надеясь на лучшее, в ранние 1980-е года исследователи стали пытаться идентифицировать специфическое генетическое нарушение, приводящее к кистозному фиброзу. После почти десяти лет борьбы, они выявили затронутый ген и точно установили мутацию, которая наиболее часто приводит к заболеванию. В то же время они могли лишь предполагать нормальную функцию гена по той роли, которую играют белки, полученные от поврежденной ДНК. Затем, в захватывающей серии открытий, исследователи выяснили, что эти белки служат в качестве каналов, через которые происходит вход и выход хлоридов, одного из компонентов соли в клетке. Они также объяснили, каким образом повреждение в гене блокирует транспорт хлоридов, и исследовали как отсутствие хлоридов приводит к явным симптомам кистозного фиброза. Эти находки способствовали возникновению новых идей в плане терапии, некоторые из которых однажды смогут излечить заболевание.
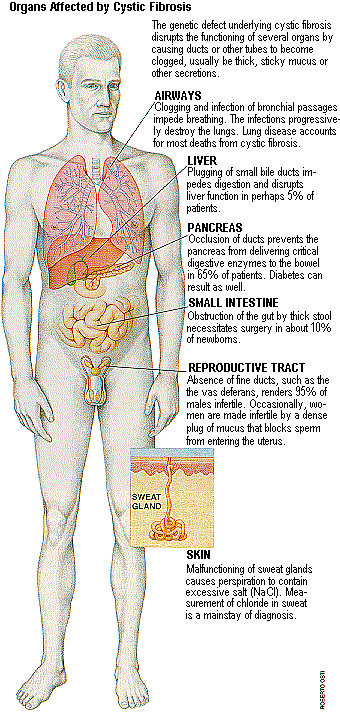
Генетический дефект, лежащий в основе кистозного фиброза , нарушает функционирование нескольких органов, вызывая засорение протоков и других трубок, которые становятся необычно толстыми , забитыми слизью или другими секретами.
Дыхательные пути. Засорение и инфекции бронхов затрудняют дыхание.
Печень. Закупоривание малых желчных протоков затрудняет переваривание и нарушает печеночную функцию у 5% пациентов.
Поджелудочная железа. Закупорка протоков не позволяет поджелудочной железе поставлять пищеварительные ферменты в кишечник у 65% пациентов. Может быть также диабет.
Тонкая кишка. Непроходимость кишечника, требующая хирургического вмешательства, примерно у 10 новорожденных.
Репродуктивные пути. Отсутствие протоков таких как vas deferans представляет 95% случаев мужского бесплодия. Порой женщина становится бесплодной вследствие блокирования плотной пробкой слизи проникновения спермы в матку.
Кожа. Нарушение потовых желез приводит к выделению пота, содержащего повышенное количество соли. Измерение хлоридов в поте - это главное доказательство диагноза.
Успехи в молекулярной области, которые ведут к многообещающему моменту в истории медицины не могли быть достигнуты без пионерских усилий врачей, многие из которых получили свое первоначальное представление о кистозном фиброзе у кровати больных. В самом деле, за прошедшие десятилетия клиническое изучение дало больше информации о заболевании, чем биохимические исследования.
Один из главных вкладов сделан в 1938 году Дороти Андерсен из Колумбийского Университета ( Dorothy H. Andersen of Columbia University). После выполнения аутопсий у младенцев и детей, и анализа медицинских историй курсантов военно-морского училища, Андерсен дала первое исчерпывающее описание симптомов кистозного фиброза и изменений, возникающих при в органах. Эти изменения, как отмечала она, почти всегда включают деструкцию поджелудочной железы (даже у младенцев) и часто инфекцию и повреждения в легких и дыхательных путях. Андерсен также дала имя болезни, назвав ее "кистозным фиброзом панкреса", на основании особенностей в панкреатической ткани, выявляемых на микроскопическом уровне(Рис.1).
Поздней в 1940-ых годах врачи дали дальнейшее описание систем протоков и других проходов в органах, наблюдаемое при кистозном фиброзе: они становятся засоренными необычно толстым секретом. В поджелудочной железе, например, протоки, которые доставляют пищеварительные ферменты к кишечнику почти всегда становятся закупоренными, снижая способность организма к измельчению пищи и извлечению питательных веществ из нее(Рис.1).
В легких бронхи и бронхиолы становятся закупоренными. Эти проходы обычно омываются тонким слоем слизи, которая ловит вдыхаемые частички и несет их к гортани для удаления. Но у пациентов с кистозным фиброзом слизь чрезвычайно толстая и слишком прочна для удаления. Это изменение само по себе сужает проходы для воздуха и затрудняет дыхание. Более того, если в дыхательных путях остаются бактерии, они становятся склонными к инфекциям. Эти инфекции, имеющие тенденцию возвращаться вредны для легочной ткани , привлекая иммунные клетки, секретирующие повреждающие вещества и ферменты. Со временем хронические инфекции постепенно разрушают бронхи и вместе с закупориванием дыхательных путей в конце концов приводят к потери дыхания (к дыхательной недостаточности) failure (Рис.1).
В 1946 году была обнаружена генетически обусловленность кистозного фиброза. После проверки типа наследования заболевания в семьях было установлено, что оно наследуется как рецессивный признак, и, возможно, обусловлено мутацией одного гена. Если младенец наследует поврежденную копию гена от обоих родителей, и, следовательно, нет нормальных молекул белка, определяемого этим геном, ребенок становится больным; однако, получение одной хорошей и одной поврежденной копий не приводит к заболеванию.
Кистозный фиброз, как известно в настоящее время, относится к наиболее обычным генетическим болезням и поражает главным образом белых. Около 5% белых американцев являются бессимптомными носителями, имеющими в своих клетках одну мутантную форму гена . В Европе один ребенок из 2500 несет две дефектные копии и имеет заболевание. В США эти числа соответствуют возникновению 1000 новых случаев в год ,и на сегодня общее количество больных составляет 30000.
Help from a Heat Wave Помощь от жары
Примерно через семь лет после установления типа наследования Нью-Йорк Сити загорал в волне тепла. Госпитали принимали непропорционально большое число детей с кистозным фиброзом, которые становились обезвоженными более быстро, чем другие курсанты военно-морского училища. Паул ди Сант'Агнесса (Paul di Sant'Agnese) со своими коллегами в Колумбийском Университете обнаружили, что мальчики и девочки с кистозным фиброзом теряли избыточное количество соли с потом. Причина повышенного выделения соли не была распознана в течение многих лет, но это наблюдение имело большую клиническую ценность. Это привело к тестированию, которое остается краеугольным камнем при диагностике: измерение содержания хлоридов в поте.
Спустя годы , такие клинические наблюдения привели к ранней более точной диагностике и лучшему лечению. Например, недостаточность поджелудочной железы на сегодня не является угрожающей жизни, т.к. пациенты могут заменить недостающие переваривающие ферменты лекарствами в капсулах, принимаемых во время еды. Тогда как проблемы пищеварения можно контролировать, поражение легких объясняет более чем 90% нетрудоспособности и смертности у пациентов с кистозным фиброзом. Выбор лечения заболеваний легких растет. Современная терапия включает старые надежные методы, называемые дренажной позой и перкуссия грудной клетки. Пациенты лежат таким образом, что их голова наклонена вниз; затем кто-нибудь мягко постукивает по его спине или грудной клетке, как если бы постукивал по дну бутылки с кетчупом, пытаясь вычистить слизь из дыхательных путей. Но пациенты так же получают пользу от антибиотиков, которые помогают контролировать повторяющиеся инфекции (хотя обычно , не уничтожая их). Около двух лет назад стало доступно и другое лечение: ингаляция лекарством, называемым ДНК-зой (DNasa). Это соединение нацелено на разрушение слизи путем переваривания длинных прямых нитей ДНК, освобождающейся из погибающих клеток.
Изучение в направлении биохимических основ кистозного фиброза прогрессировало более медленно, чем в клинической работе , но более интенсивно продвинулось в первую половину 180-ых годов. В это время ученые пришли к пониманию, что нарушение функций эпителиальной ткани привело к дефектам в каждом органе, пострадавшем при кистозном фиброзе (Эпителий это лист клеток, которые формируют барьер между разными компартментами тела; такие листы, которые часто секретируют слизь, выстилают кишки и многие протоки.) В частности, по двум путям исследований было обнаружено, эпителий пациентов с кистозным фиброзом сравнительно плохо проницаем для хлоридов. Это открытие означает, что некоторые функции хлорид-транспортных канальцев в эпителиальной ткани должны быть нарушены.
В одном ряду таких исследований, Паула М. Квинтона из Университета в Калифорнии на Риверсайд( Paul M. Quinton of the University of California at Riverside) было найдено, что эпителий, выстилающий протоки потовых желез, не способен эффективно принимать хлориды из полости, или просвета желез. Эта находка объяснила, почему люди с кистозным фиброзом имеют необычно соленый пот. Обычно пот продуцируется в основании потовых желез; затем вытекает на поверхность кожи через тонкий проток. Сначала пот это раствор, богатый ионами натрия и хлора, компонентов соли. Но по мере того, как жидкость проходит через проток, ионы всасываются эпителием и покидают воду. Таким образом, пот появляется для охлаждения поверхности кожи и, лишь слегка солоноват. У пациентов с кистозным фиброзом неспособность эпителиальной ткани к поглощению и, следовательно неспособность к поглощению натрия из просвета протока приводит к тому, что пот остается с избытком натрия и хлора и становится ненормально соленым(Рис.1).
В другой серии работ, Михаэла Р. Наулеса и Ричарда С. Баучера из Университета в Северной Каролине на Чапел Хилл( Michael R. Knowles and Richard C. Boucher of the University of North Carolina at Chapel Hill ) были исследованы легкие. Авторы обнаружили, что движение хлоридов от эпителиальной ткани в просвет дыхательных путей уменьшено, и что поглощение эпителием натрия увеличено. Уменьшенный транспорт хлоридов продемонстрирован теперь также и в эпителии протоков поджелудочной железы у мыши и в кишечнике у пациентов. ducts in mice and of
Finally, the Gene Is Found Ген найден
По мере развития работ по транспорту хлоридов многие ученые начали интенсивные работы по поиску гена, ответственного за возникновение кистозного фиброза. Кульминация этих усилий пришлась на 1989 год, когда большая группа сотрудников , возглавляемых Лэп-Чи Цу (Lap-Chee Tsui) и Джоном Р. Райрденом ( John R. Riordan) из Госпиталя для ослабленных детей в Торонто( Hospital for Sick Children in Toronto) и Френсисом С. Коллинзом( ( in , 1998), и затем в Университете в Мичигане и который теперь является директором проекта Геном Человека в NIH, объявили, что ген выделен. Зная, что белковый продукт гена вероятно влияет прямым или непрямым образом на передвижение хлоридов , они назвали белок регулятором трансмембранного переноса при кистозном фиброзе [cystic fibrosis transmembrane conductance regulator (CFTR)]. B процессе исследования гена были идентифицированы аномалии в ДНК, которые, по-видимому, объясняют около 70% случаев возникновения кистозного фиброза. Абревиатура AF508 обозначает мутацию, которая является делецией трех нуклеотидов (строящих ДНК блоков) гена. Такая потеря приводит в возникновению белкового продукта, лишенного единственной аминокислоты: фенилаланина в позиции 508(Рис.2).
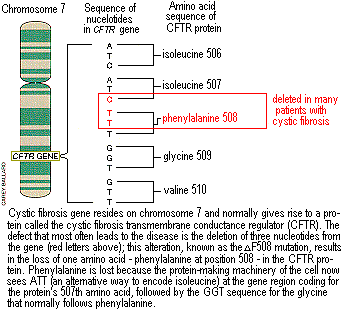
Дефект, который наиболее часто ведет к заболеванию - делеция в гене трех нуклеотидов; это нарушение, известное как мутация дельтаF508, приводит к потере аминокислоты фенилаланина в позиции 508 белка CFTR. Потеря фенилаланина происходит потому, что теперь триплет АТТ кодирует 507-ю аминокислоту, за которой следует последовательность ГГТ, кодирующая глицин, следующий в норме за фенилаланином.
Было захватывающее сообщение для всех, кто интересуется кистозным фиброзом; оно обещает открыть новые перспективы в понимании и новый выбор для терапии. Тем не менее исследователи искали дополнительных доказательств, которые бы уточнили выделение гена. Сильное подтверждение могло быть получено при встраивании здоровой версии в клетки от пациентов с кистозным фиброзом, и, следовательно , коррекции дефекта транспорта хлора. Тщетно, было трудно сконструировать даже направленную версию гена. Летом 1990 года, однако, наш коллега Ричард Дж. Грегори (Richard J. Gregory) из Genzyme Corporation решил эту проблему.
Двое из нас и наши сотрудники не теряли времени, пытаясь вставить ген в эпителиальные клетки, взятые из дыхательных путей больных кистозным фиброзом. Genzyme Corporation Мы подвергали клетки действию цАМФ, молекуле, которая в норме стимулирует транспорт хлоридов в эпителии дыхательных путей, но не действует на ткань от пациентов с кистозным фиброзом. Мы были поражены, увидев, что цАМФ теперь вызывает вытекание хлоридов из обработанных клеток; ген вероятно делал клетки нормальными. Мы не были одиноки в своем восхищении. Коллинз и ряд его коллег получили подобные результаты, используя другие методы в эпителиальных клетках поджелудочной железы.
Успехи культивирования клеток наводили на мысль, что доставка здоровых генов CFTR может исправить их лежащую в основе биохимическую аномалию, давая ложную надежду. Но мы также знали, как будет видно, что существуют многие препятствия достижению этой цели. Тем временем стали вырисовываться другие проблемы в данной области: как белок CFTR точно влияет на движение хлоридов.
What Does This Protein Do?Что делает этот белок?
Линейная последовательность аминокислот, которую просто вывести раз ген выделен, предлагает несколько непосредственных ключей для нормального поведения белков. Примечательно, что последовательность очень похожа на последовательность, обнаруженную у семейства белков, называемых движущими АТФазами или АВС- транспортерами (потому, что они несут то, что называется АТФ связывающими кассетами). Предположили, что CFТR по поведению и упаковке, трех-мерной структуре может также напоминать семейство.
Семейство движущих АТФаз включает ряд белков, используемых бактериями для накачивания питательных веществ через свою клеточную мембрану; это семейство включает также лекарство-резистентный белок, который изгоняет химиотерапевтические лекарства из клеток опухоли [см. "Multidrug Resistance in Cancer," by Norbert Kartner and Victor Ling; SCIENTIFIC AMERICAN. March 1989]. В упакованном состоянии упомянутые АТФазы как правило имеют четыре главные структурные части, или домена: два, которые простираются на мембрану (каждый из которых содержит несколько трансмембранных сегментов) и два, которые находятся в цитоплазме. Последние две единицы, известные как нуклеотид-связывающие домены, забирают и расщепляют АТФ (нуклеотид аденозин трифосфат) чтобы получить энергию, необходимую для накачивания. Молекула CFTR, как было предсказано, принимает подобную форму и, как увидим, имеет дополнительный компонент, находящийся в цитоплазме.
Основываясь на активности АТФаз, некоторые исследователи выдвинули предположение, что CFTR является АТФ- управляемой помпой, которая активно транспортирует некоторые вещества внутрь и из клеток эпителия; транспортируемые вещества затем индуцируют хлоридный транспорт через клеточную мембрану по отдельному канальцу. Они предложили эту сложную схему, потому что не знали о ионных канальцах(таких, которые должны быть необходимы для более направленного движения хлоридов), напоминающих предсказанную структуру упаковки CFTR.
Согласно второй гипотезе предположили, что CFTR сам присоединяется к хлоридным канальцам, влияя на их активность. Согласно третьей гипотезе предположили, что СFTR может прямо служить хлоридным канальцем даже несмотря на то ,что его структура необычна для любого ионного канальца, распознаваемого при испарении. По такому сценарию два соединенных с мембраной домена должны формировать пору, через которую ионы хлора проникают сквозь мембрану.
По мере продвижения исследований данные подтвердили третью идею: CFTR формирует канал из собственных белков . Мы обнаружили, что перенос гена CFTR в непроницаемые для хлора клетки придает им способность к перемещению иона. Если ген был изначально изменен таким образом, что затрагивал части белка CFTR, который, как полагают, помогает передвигать хлориды через каналец, то сродство канальцев к хлоридам уменьшается; этот эффект был продемонстрирован нашим коллегой Мэтью П. Андерсоном из Университета в Айове (Matthew P. Anderson of the University of Iowa). Последние сомнения рассеялись, когда Райрден (Riordan) со своими коллегами вставил высоко очищенный белоки CFTR в искусственные клеточные мембраны ( липидные бислои), не содержащие никаких других канальцеподобных белков. Добавление белков позволило ионам передвигаться сквозь мембрану.
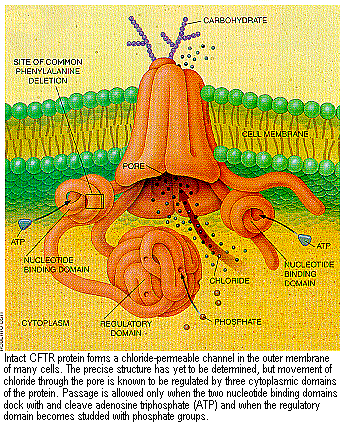
Последующие исследования прояснили функцию "экстра" компонента CFTR, не найденную в движущих АТФазах. На основании определенных коротких последовательностей (оn the basis of certain short sequences) в пределах этого компонента таинственный сегмент был дедуцирован как регуляторный домен R, активность которого в цитоплазме контролируется увеличением или уменьшением количества фосфатных групп. Ряд экспериментов, включая опыты наших коллег Сенг Г. Ченг из Университета в Айове( Seng H. Cheng of Genzyme and Devra P. Rich of the University of Iowa),показали, что когда домен R лишен фосфатных групп, ионы хлора не могут протекать внутрь поры канальца. Но при химических изменениях в клетке(особенно при увеличении уровня циклического АМФ), которые с помощью ферментов обеспечивали домен фосфатами, происходило дополнительное стимулирование передвижения хлоридов через пору.
Полезно, несмотря на простоту, представлять себе, что когда регуляторный домен не фосфорилирован, он ведет себя подобно входному блокатору цитоплазматического отверстия мембранной поры. Добавление фосфатов каким-то образом переставляет домен(открывая вход), позволяя ионам хлора проникать внутрь поры. Другие исследования продемонстрировали, что нуклеотид- связывающие домены также влияют на активность канальца. Для того, чтобы ионы двигались через пору, эти домены должны связывать и, вероятно, расщеплять АТФ.
How the Mutations Make Mischief Как мутации делают повреждения
Известно, что белок CFTR формирует хлоридный каналец
и существует несколько идей о том как
передаются молекулярные функции, важном
вопросе, на который еще нужно ответить: а
именно как мутации гена CFTR приводят к
отсутствию хлоридного транспорта? Эффект
наиболее обычной мутации ДНК, делеции,
которая приводит к потери фенилаланина 508
из белка CFTR изучен наиболее хорошо. Эта делеция порождает
то, что известно как дефект внутриклеточного передвижения (trafficking). Многие белки, среди которых
и нормальная молекула СFTR, подвергаются
процессингу после синтеза. Они приобретают
некоторые группы сахаров в клеточном
компартменте, называемом эндоплазматическим ретикулумом, после
которого они забирают сахар в аппарате
Гольджи до отправления к клеточной
мембране. Мутантный белок в отличие от
нормального не способен покидать
эноплазматический ретикулум. Его
перемещение останавливается
предположительно из-за системы контроля
качества в эндоплазматическом ретикулуме,
распознающей, что белок упакован
неправильно. Белки, идентифицированные как
дефектные , скорее для деградируют, чем
дальше продолжают процессинг.
Несмотря на то, что мутация фенилаланина 508
наиболее обычна, у людей с кистозным
фиброзом сейчас идентифицированы сотни
других. Как и для мутации 508, многие из этих
изменений блокируют белки на пути к
клеточной мембране. Некоторые мутации
полностью блокируют создание белка, другие
позволяют белку продуцироваться и
вставляться в клеточную мембрану, но
препятствуют молекуле CFTR правильно оперировать.В
последнем случае мутации могут
предупреждать передвижение хлоридов ,
нарушая функцию нуклеотид связывающего
домена или введение потока внутрь ион-транспортирующей
поры(Рис.3).
Вообще, люди, чьи клетки несут две копии мутантного гена фенилаланин 508 имеют предрасположенность к тяжелому заболеванию, вероятно мало если и есть таких молекул белка, которые избегают застревания в эндоплазматическом ретикулуме. У людей, чьи гены позволяют достичь по крайней мере нескольким CFTR клеточной мембраны, они позволяют до некоторой степени транспортировать хлориды. Остаточная активность до некоторой степени облегчает тяжесть симптомов. Однако предсказания в каждом индивидуальном случае остается проблематичным. В самом деле, Два пациента с одинаковой мутацией в обеих копиях гена CFTR могут значительно отличаться по степени нарушений органов, которыми они страдают. Это расхождение возникает потому, что на течение болезни вероятно влияют другие генетические и внешнесредовые факторы, остающиеся слабо изученными.
Скромно отметим, что развивающееся понимание генетических дефектов еще не полностью объясняет, как нарушения транспорта хлоридов в эпителии легкого изменяет транспорт натрия ,и каким образом эти изменения приводят к накоплению слизи в бронхах. Обнаружено, что субмукозные железы, вырабатывающие слизь, которая лежит на поверхности эпителия , продуцируют большое количество белка CFTR. Какую роль эти железы играют при заболевании? Ученые были озадачены фактом, что дыхательные пути пациентов с кистозным фиброзом расположены к инфицированию некоторыми бактериями в большей степени, чем другими. Например, инфекции Pseudomonas aeruginosa и Staphylococcus aureus особенно часты. Понимание, почему разрастаются определенные организмы начинает появляться только теперь.
Исследователи удивлены также тем, что белок CFTR обладает функциями кроме его роли в хлоридных канальцах. Среди возможных вариантов рассматривается такой, что CFTR может помогать регулировать хлоридные канальцы, отличные от CFTR. Установлено также, что молекула может изменять или смешивать сахара на поверхности эпителия таким образом, что создаются условия для заселения определенными бактериями.
В сообщении от 5 января 1996 г. , вышедшем в , представлены доказательства, что , белок CFTR играет роль в поглощении бактерий Рseudomonas aeruginosa эпителиальными клетками легких. Эпителиальные клетки от CF пациентов, гомозиготных по мутации F508 не глотают эти бактерии, и такие пациенты становятся гиперчувствительными к хронической легочной инфекции.
Strategies for Treatment Стратегии лечения
Несмотря на вопросы, оставшиеся без ответа, знания, накопленные с 1989 г. наводят на мысль о нескольких путях для борьбы с кистозным фиброзом. Первый - компенсировать потерю CFTRхлоридного канальца путем увеличения активности хлоридных канальцев другого класса. Например, с помощью канальцев, контролируемыми ионами кальция, которые как известно существуют на поверхности эпителиальных клеток, обращенной к просвету. Эти молекулы не могут нейтрализовать потерю канальца CFTR, но возможно их проводимоть хлоридов можно увеличить искусственно. Эта возможность проверяется на пациентах.
Однажды врачи смогут доставлять очищенные белки CFTR к клеткам, нуждающимся в них. Работы на клетках в культурах показали, что молекулы белка могут корректировать поток хлоридов в клетках, несущих мутантный ген CFTR. В теории, возможна другая тактика - вводить лекарства, способные сопровождать мутантные молекулы CFTR из эндоплазматического ретикулума через аппарат Гольджи в клеточную мембрану. Эта идея представляется ценной, т.к. мутантные AF508 белки CFTR, которые остаются в эндоплазматическом ретикулуме, при экспериментальном встраивании их в наружную мембрану клеток обычно функционируют довольно хорошо. В настоящее время, однако, мы не знаем никаких лекарств, которые могли бы исправлять аномалии внутриклеточного перемещения. Другое направление, еще не проверенное, - использование лекарств для увеличения активности любого мутантного канальца, позволяющим ему найти свою дорогу к клеточной мембране.
Выбор лечения, привлекающий наибольшее внимание, однако, - это генотерапия, цель которой - доставить нормальную копию гена CFTR, нуждающимся в ней клеткам. Если все идет хорошо, ДНК, включившаяся в клетки мишени , управляет синтезом нормального белка CFTR и отменяет первичную биохимическую аномалию, являющуюся источником возникновения кистозного фиброза. Введение гена является приоритетным направлением потому, что при этом происходит замена всех функций белка CFTR, включая и те, которые еще не распознаны.
Лучше всего изученный метод генотерапии использует способность вирусов проникать в клетки, принося в них свою ДНК. Мы и другие исследователи обращают особое внимание на аденовирусы в качестве генных носителей, или векторов потому, что они естественно способны инфицировать дыхательные пути человека, но как правило вызывают относительно безвредную болезнь так же, как обычный холод. Аденовирусы изменяют двумя способами: удаляют определенные вирусные гены для предотвращения их размножения в клетке, приводящего к возникновению симптомов. И удаленную ДНК замещают нормальным геном CFTR . Нашей группой, а также группой Рональда Г. Крайстала ( Ronald G. Crystal),затем в the National Heart, Lung и Институте крови(Blood Institute), и Джеймсом М. Уилсоном (James M. Wilson), затем в Университете в Мичигане (the University of Michigan), было продемонстрировано, что такие векторы могут доставлять ген CFTR культивируемым эпителиальным клеткам и клеткам дыхательных путей животных. Более того, клетки используют ДНК для синтеза молекул CFTR, которые функционируют как здоровые хлоридные канальцы.
На основании такого рода опытах несколько исследовательских групп начали попытки доставлять ген CFTR пациентам, используя векторы , генетически сконструированные из аденовирусов. Цель этих ранних экспериментов была сначала - оценка безопасности. Равно , мы и другие также тестировали способность CFTR несущих аденовирусов корректировать транспорт хлоридов в эпителии носа у пациентов. Мы выбрали эпителий носа, т.к. он подобен эпителию в бронхах, но более доступен.
Наши первые результаты были ободряющими. В целях эксперимента мы прикладывали измененный вирус прямо к маленькому пятну эпителия в носу. Воздействие частично корректировало транспорт хлоридов на время. Затем, однако, наша работа была менее успешна, и одной из других групп не было зарегистрировано увеличения потока хлоридов. Эти находки показали, что аденовирусные векторы нуждаются в усовершенствовании , чтобы быть пригодными в качестве агентов доставки гена при терапии.
Даже если бы нашли пути увеличения эффективности доставки гена вирусом, оставались еще другие трудности. Большинство клеток эпителия заменяются в течение нескольких месяцев. Следовательно, генотерапию вероятно нужно применять несколко раз в год, по крайней мере до тех пор, пока не смогут быть индуцированы разреженные, долго живущие клетки, которые дадут возможность для замещения клеток , позволяющих принять надолго нормальный ген CFTR. Кроме неудобства и дороговизны, необходимость для множественных введений касается человеческой реакции на аденовирусы посредством увеличения иммунного ответа, который элиминирует их и предотвращает повторное инфицирование. Для успешного проведения генотерапии , исследователи нашли путь к "укрытию" аденовирусов от иммунной системы. или к созданию вирусных или других векторов, которые не вызывают иммунного ответа.
Существует привлекательная альтернатива - передавать посредством вирусов, которые должны быть покрыты, терапевтический ген с жировыми молекулами , которые не узнает иммунная система, но которые тем не менее способны вводить ДНК в клетки. Недавно Эриком Олтоном (Eric Olton) и его сотрудниками в Royal Brompton Hospital в Лондоне были проведены на человеческих пациентах исследования, которые наводят на мысль, что это направление позволяет восстановить проницаемость дыхательных путей для хлоридов эпителия , хотя его группа, как и мы , изучали до сих пор только назальную ткань. Более того, доставку генов с помощью невирусных систем необходимо сделать более эффективной. Исследователям предстоит еще многое изучить для точного представления, как потеря белка CFTR приводит к проявлениям кистозного фиброза. И должно быть преодолено множество технических проблем до того момента, когда терапия сможет компенсировать эту потерю рутинно. Тем не менее, прогресс сделан на многих фронтах. Трудно не быть оптимистичным, что идущая работа даст более совершенную терапию в ближайшие несколько лет.
Testing Dilemmas Трудности тестирования
Теперь, когда многие генетические мутации, приводящие к кистозному фиброзу, уточнены будущие родители могут просто выяснить, являются ли они носителями заболевания, т.е. являются ли их клетки молчащими носителями дефектной копии гена CFTR. Пара может также установить, унаследовал ли уже развивающийся плод две измененные копии гена (одного от каждого родителя) и болен кистозным фиброзом.
Для многих людей трудно решить, как быть дальше, если они получили результат тестирования , указывающий на их рецессивное носительство. Возникает тревога отчасти потому, что лаборатории ,выполнившие генетический анализ, не могут определить каждую мутацию гена CFTR. Следовательно, успокаивающая отрицательная находка не полностью исключает, что кто-то является носителем или кто-то страдает кистозным фиброзом. (Результаты более подходящего пренатального теста будут заключительными, однако, если показано, что плод лишен специфического мутантного гена CFTR, который как показано, несут его родители.) Более того, еще невозможно предсказать степень выраженности симптомов у лица, которое унаследовало два мутантных гена CFTR; даже если унаследованные гены обычно ассоциируют с тяжелым или менее тяжелым заболеванием, такие ассоциации не обязательно содержат истинное понимание в каждом индивидуальном случае.
Некоторые пары могут слишком поспешно полагать, что исследование будет прогрессировать достаточно быстро ,чтобы защитить детей, родившихся сегодня, от угрожающих жизни нарушениях в легких, характерных для кистозного фиброза. Медицинские исследования все еще часто наталкиваются на неожиданные препятствия и претерпевают задержку до того, как достигнут своей конечной цели. С этих пор, несмотря на то, что лечение будет становиться более эффективным возможно отмечено так в наступающие годы, но никто не может предсказать точно, когда с кистозным фиброзом станет проще справиться . Будущим родителям необходимо понять, что сегодня при рождении ребенку, родившемуся с кистозным фиброзом еще предстоит справиться с заболеванием, и его может не пощадить преждевременная смерть.
Из-за таких неопределенностей решение представляется предельно сложной задачей. Это волнующий период в изучении кистозного фиброза, но попытки пар-энтузиастов застревают в разрыве между сегодняшней технологией и предвкушаемым успехом, который еще не стал реальностью.
from Scientific American, December, 1995 Vol. 273: pp. 52-59
Further Reading
- CYSTIC FIBROSIS: MOLECULAR BIOLOGY AND THERAPEUTIC IMPLICATIONS. Francis S. Collins in Science, Vol. 256, pages 774-779; May 8, 1992. CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR: A CHLORIDE CHANNEL WITH NOVEL REGULATION. M. J. Welsh, M. P. Anderson, D. P. Rich, H. A. Berger, G. M. Denning, L. S. Ostedgaard, D. N. Sheppard, S. H. Cheng, R. J. Gregory and A. E. Smith in Neuron, Vol. 8, No. 5, pages 821-829; May 1992.
- THE CYSTIC FIBROSIS TRANSMEMBRANE CONDUCTANCE REGULATOR. J. R. Riordan in Annual Review or Physiology, Vol. 55, pages 609-630; 1993.
- MOLECULAR MECHANISMS OF CFTR CHLORIDE CHANNEL DYSFUNCTION IN CYSTIC FIBROSIS. M. J. Welsh and A. E. Smith in Cell, Vol. 73, No. 7, pages 1251-1254; July 2,1993.
- CYSTIC FIBROSIS. M. J. Welsh, L. C. Tsui, T. F. Boat and A. L. Beaudet in Metabolic and Molecular Basis or Inherited Disease. Edited by C. R. Scriver, A. L. Beaudet, W. S. Sly and D. Vade. McGraw-Hill, 1994.