. Эту стадию можно назвать “burnt-out” («сгорания»), иногда она длятся недели, а иногда месяцы. Ребенок страдает умственной отсталостью, слепотой. Теряется контакт с окружающими. Больные редко живут более 2-3 лет.
ПатологияНейропатология глобоидно-клеточной лейкодистрофии настолько уникальна, что диагноз не вызывает затруднения в том случае, если для исследования доступна мозговая ткань. Патология ограничивается главным образом нервной системой. На терминальной стадии миелин утрачен почти полностью, за исключением (в некоторых случаях) субкортикальных intergyral arcuate волокон. При микроскопии выраженная нехватка миелина с некоторой дегенерацией аксонов наблюдают во всем мозге. Обширный фибриллярный глиоз и инфильтрация многочисленных макрофагов, чаще многоядерных («глобоидные клетки») являются специфическими признаками. Избыточное количество глобоидных клеток присутствует в области активной демиелинизации и они часто группируются вокруг кровеносных сосудов. Олигодендроциты исчезают быстро. Глобоидные клетки содержат PAS –позитивные трубчатые (тубулярные) и нитевидные включения с полигональными поперечными сечениями, которые по своей структуре идентичны химически чистому галактозилцерамиду. Периферические нервы часто сильно увеличены и уплотнены с выраженным эндонейральным фиброзом, сегментарной демиелинизацией и процессом ремиелинизации. Эндонейральные макрофаги и Шванновские клетки содержат тубулярные включения сходные с включениями в глобоидных клетках белого вещества мозга.
Биохимия. У всех известных до сегодняшнего времени пациентов с болезнью Краббе наблюдали дефицит активности галактозилцерамидазы (РИС.1).
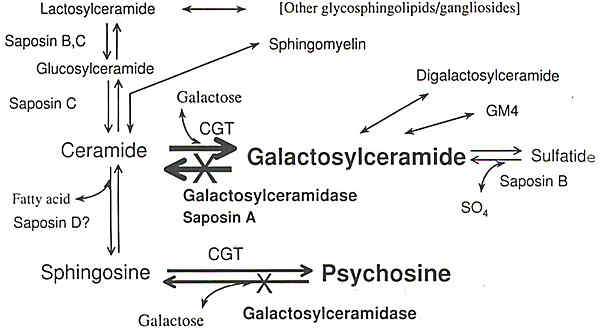 Рис.1. Metabolic pathways pertinent to galactosylceramide and related compounds. In the synthetic pathway, sphingosine is first acylated to ceramide, which in turn is galactosylated by UDP-galactose: ceramide galactosyltransferase (СПЕ) to form galactosylceramide. The same enzyme can galactosylate sphingosine directly to generate psychosine. Both galactosylceramide and psychosine are degraded by galactosylceramidase, which is genetically deficient in Krabbe disease. In vitro degradation of galactosylceramide requires in addition to the enzyme, a shingolipid activator protein, saposin A. Galactosylceramide is further sulfated to form sulfatide. Both galactosylceramide and sulfatide are characteristic myelin glycolipids.
Рис.1. Metabolic pathways pertinent to galactosylceramide and related compounds. In the synthetic pathway, sphingosine is first acylated to ceramide, which in turn is galactosylated by UDP-galactose: ceramide galactosyltransferase (СПЕ) to form galactosylceramide. The same enzyme can galactosylate sphingosine directly to generate psychosine. Both galactosylceramide and psychosine are degraded by galactosylceramidase, which is genetically deficient in Krabbe disease. In vitro degradation of galactosylceramide requires in addition to the enzyme, a shingolipid activator protein, saposin A. Galactosylceramide is further sulfated to form sulfatide. Both galactosylceramide and sulfatide are characteristic myelin glycolipids.
Галактозилцерамидаза является гидролитическим ферментом с кислым рН оптимумом, локализованным в лизосомах. Поэтому концептуально заболевание входит в категорию лозосомных болезней, охарактеризованных Hers (1966). По существу все лизосомные болезни являются «болезнями накопления», при которых субстрат генетически дефектного фермента накапливается с аномально высоким уровнем. Этот фермент специфичен для определенных гликолипидов с терминальным галактозным компонентом при
β аномерной конфигурации. Главным субстратом фермента является галактозилцерамид, который локализуется почти исключительно в миелиновой оболочке. Другими субстратами фермента являются психозин (psyhosine или galactosylsphingosine), monogalactosyldiglyceride и предшественник seminolipid (1-alkil,2-acyl-, 3-galactosyl glycerol). Для деградации этих субстратов
in vivo кроме фермента галактозилцерамидазы необходим активирующий белок – saposin A. Уникальной биохимической характеристикой болезни Краббе является
отсутствие аномального накопления галактозилцерамида в мозге, что противоречит тому, что можно было бы ожидать при таком энзиматическом дефекте. Это парадоксальное явления можно было бы объяснить исключительной локализацией галактозилцерамида в миелиновой оболочке и очень быстрым и ранним исчезновением миелинизирующих клеток во время течения заболевания. Поскольку очень быстрое исчезновение миелинизирующих клеток устраняет источник синтеза галактозилцерамида, то он не накапливается выше уровня, который достигается на ранних стадиях миелинизации. Однако родственный токсический метаболит психозин (galactosylsphingosine) накапливается до аномального уровня и рассматривается как ключевой компонент в патогенезе этого заболевания. Галактозилцерамид обладает уникальной способностью вызывать инфильтрацию глобоидных клеток при имплантации его в мозг. Такие экспериментально индуцированные глобоидные клетки морфологически выглядят также как и клетки пациентов с болезнью Краббе.
Патогенез классической инфантильной GLD. Наиболее типичными патологическими признаками болезни Краббе являются:
1) инфильтрация макрофагов, которые чаще всего многоядерны и содержат PAS-позитивные вещества («глобоидные клетки»);
2) быстрое и почти полное исчезновение олигодендроцитов;
3) отсутствие аномального накопления первичного субстрата дефектного фермента галактозилцерамида в противовес тому, что ожидается при болезнях накопления из-за генетического дефекта фермента деградации.
Эти фенотипические характеристики должны быть объяснены как следствие лежащего в их основе генетического дефекта. Дефектная деградация двух субстратов – галактозилцерамида и психозина (galactosylsphingosine), играет критическую роль в патогенезе заболевания. Несмотря на фундаментальные различия этих механизмов, они тесно переплетаются при формировании уникального фенотипа этого заболевания.
Глобоидные клетки Очевидно, что нарушенная деградация галактозилцерамида является главным фактором, определяющим основной патологический признак заболевания, т.е. глобоидные клетки. Давно известно, что свободный галактозилцерамид обладает специфической способностью обеспечивать инфильтрацию макрофагов в мозг.
Пока неизвестно, обладают ли другие агенты такой же способностью
in vivo. Попав в мозг, макрофаги подвергают фагоцитозу галактозилцерамид и трансформируются в многоядерные глобоидные клетки. Морфологически включения в глобоидных клетках идентичны самому галактозилцерамиду. Реакция глобоидных клеток может быть реконструирована (восстановлена) следующим путем. Во время начала активного периода миелинизации начинается и преобразования миелиновой оболочки. Однако в мозге больных галактозилцерамид не распадается из-за дефекта галатозилцерамидазы. Свободный галактозилцерамид таким образом генерирует инфильтрацию гематогенных макрофагов, которые становятся PAS –позитивными, часто многоядерными глобоидными клетками.
Психозиновая (psychosin) гипотеза. Значительную рано возникающую деструкцию миелин-формирующих клеток трудно объяснить присутствием галактозилцерамида, поскольку не существует никаких экспериментальных доказательств того, что галактозилцерамид относится к метаболическим токсинам. Галактозилцерамид, имплантированный в мозг, не вызывает дисфункций, кроме глобоидно-клеточной реакции. С другой стороны, близкородственный метаболит психозин (галактозилсфингозил) высоко цитотоксичен и вызывает быстрый и фатальный геморрагический инфаркт при имплантации в мозг. В тканях млекопитающих психозин генерируется только галактозилированием сфингозина (sphingosine) галактозилцерамид синтазой (galactosylceramide synthase), UDP-galactose: ceramid galactosyltransferase (СGT), но не де-ацилированием галактозилцерамида. Поскольку CGT почти исключительно локализована в миелин-образующих клетках, то синтез психозина происходит только в олигодендроцитах и Шванновских клетках. Психозин определяется в нормальном мозге высокочувствительными аналитическими методами, но его концентрации крайне малы (менее 10 picomoles/mg белка) Оказывается, он является метаболическим конечным продуктом, который в норме сразу разрушается галактозилцерамидазой. Следовательно, у пациентов с болезнью Краббе психозин не может быть деградирован.
«Психозиновая гипотеза» была впервые предложена на основе энзимологических соображений и затем его аномальное (психозина) накопление было аналитически продемонстрировано в мозге больных, а также в мозге модельных собак и мышей. Согласно психозиновой гипотезе при глобоидно-клеточной лейкодистрофии деградации не подвергается не только первичный субстрат дефектного фермента (галактозилцерамид), но и токсический метаболит галактозилсфингозил (психозин) и что впоследствии аномальное накопление психозина вызывает крайне быстрое разрушение миелин-образующих клеток. Вначале данная гипотеза была встречена со скептицизмом, но, тем не менее, не была отвергнута в течение 30 лет. Фактически, основная предпосылка гипотезы распространилась на другие сфинголипидозы (Hannun Y. et al. 1987). Однако по разным причинам эта гипотеза оказалась неприемлемой для других заболеваний также как и для GLD, за исключением нейропатической формы болезни Gaucher и болезни Neimann-Pick типа А.
Современные исследования.
Saposin A- дефицитные мыши . Экспериментальное создание новых мышиных моделей глобоидно-клеточной лейкодистрофии, обусловленное генетическим дефицитом saposin A (
in vivo галактозилцерамидазным активирующим белком) усложнило картину и заставило пересмотреть взгляды в отношении патогенеза GLD (Matsuda J. et al. 2001). Крайне важно то, что с помощью этой модели было установлено, что дефект гена галактозилцерамидазы является не единственной возможной генетической причиной, лежащей в основе глобоидно-клеточной лейкодистрофии. Кроме того, с помощью этой модели было установлено, что два патогенетических механизма – инфильтрация глобоидных клеток, обусловленная галактозилцерамидом, и утрата олигодендроцитов, обусловленная токсичностью психозина, могут оперировать независимо друг от друга. Saposin A-дефицитные мыши были созданы экспериментально посредством интродуцирования аминокислотной замены (C106F) в saposin A домене посредством
Cre/loxP системы, которая элиминировала одну из трех консервативных дисульфидных связей, важных для функциональных свойств saposins. У человека эквивалентная мутация в четвертом цистеине (to phenylalanine) в saposin C вызывает специфическую недостаточность saposin С. А мутация в пятом цистеине (to serine) в saposin B приводит к специфическому дефициту saposin B. У saposin A дефицитных мышей развивается медленно прогрессирующий паралич задних конечностей с клиническим началом около 2.5 мес и выживаемостью животных до 5 мес. Тремор и сотрясение (тела или головы), выраженные при других миелиновых мутациях, не проявлялись у этих мышей до терминальной стадии. Самцы и самки saposin A-дефицитных мышей фертильны и самки способны выращивать детенышей до трех раз. Однако во всех отношениях патология (особенности периферических нервов, демиелинизация, реактивный астроглиоз, инфильтрации глобоидных клеток, содержащих характерные включения) и биохимия (аномалии деградации субстратов галактозилцерамидазы в мозге почках и тестикулах), качественно идентичны, но в целом гораздо менее выражены у этих моделей по сравнению с twitcher мышами – природной моделью GLD, обусловленной дефицитом галактозилцерамидазы. Накопление психозина в мозге этих мышей было лишь в два раза выше нормы, тогда как у мышей twitcher он был увеличен в 10-20 раз. Saposin A-дефицитные мыши четко показали, что saposin A обязателен для активирования галактозилцерамидазы
in vivo, т.е. нормальные клеточные функции без него осуществляться не могут.
Улучшение фенотипа saposin A-дефицитных мышей во время беременности
Во время разведения мышей было замечено, что у saposin A-дефицитных беременных самок улучшались неврологические симптомы по сравнению с пораженными, но не беременными самками. Патологические признаки глобоидно-клеточной лейкодистрофии – демиелинизация с инфильтрацией глобоидных клеток – практически исчезали. Было также установлено, что у беременных saposin A-дефицитных самок была значительно снижена экспрессия (down-regulated) генов MCP-1 и TNF-α. Кроме того, у saposin A-дефицитных мышей наблюдали интенсивную экспрессию эстрогеновых рецепторов (ER-α и ER-β) на глобоидных клетках, активированных астроцитах и микроглии в демиелинизированной области. При последующей имплантации saposin A-дефицитным мышам 17 β-estradiol (E2) таблеток через определенные промежутки времени на протяжении 30-90 дней, их состояние значительно улучшалось. Эти данные подтверждают, что высокий уровень эстрогена во время беременности является одним из важных факторов в защитном эффекте беременности.
Апоптоз
Хотя в настоящее время известно, что цитотоксичность психозина, вероятно, ответственна за характерное быстрое исчезновение олигодендроцитов, точный механизм его действия пока не определен. Предполагают, что апоптоз приводит к уменьшению числа олигодендроцитов. Во всяком случае, было установлено, что число олигодендроцитов у twitcher мышей увеличивалось при подавлении процесса апоптоза. Это согласуется с наблюдениями
in vivo, когда было показано, что психозин является потенциальным индуктором апоптоза, как и С-6 церамид.
Участие иммунного и воспалительного процессов
Известно, что при прогрессировании демиелинизации увеличивается число GFAP-, Mac-1 и F4/80-позитивных клеток в периферической и центральной нервных системах мышей twitcher. Некоторые Mac-1-позитивные клетки экспрессировали также MHC класса II (Ia). Появление Ia экспрессирующих клеток совпадало с началом демиелинизации. Число Ia-иммунореактивных клеток постепенно увеличивались в областях демиелинизации, достигая плато между 30 и 40 днями после рождения в головном мозге и затем быстро снижалось, несмотря на продолжающуюся демиелинизацию. Однако в спинном мозге число Ia иммунореактивных клеток не снижалось до 50-дневного возраста. Эти наблюдения свидетельствуют либо о специфическом участии иммунологических факторов в патогенезе наследственных заболеваний, сопровождающихся демиелинизацией, либо о не специфической реакции на дегенерирующие тканевые компоненты, такие как миелин. Скрещивания twitcher мышей с MHC класса II нокаутными мышами дало возможность получить twitcher мышей с дефицитом молекул MHC класса II. У этих мышей клинические признаки и гистопатология мозга и области мозжечка были выражены слабее, чем у мышей twitcher с MHC-II позитивным бэкграундом, однако никаких улучшений патологических признаков у этих мышей в спинном мозге не наблюдали. Предварительный анализ уровня психозина также указывает на не столь высокое его накопление, чем у MNC-II-позитивных twitcher мышей, что согласовывалось с меньшей степенью патологии у этих животных. Интерлейкины, цитокины и их рецепторы также были изучены у twitcher мышей для оценки участия воспалительного процесса в патогенезе GLD. Описанное ранее улучшение фенотипа saposin A-дефицитных самок во время беременности возможно связано с противовоспалительной способностью эстрогена.
Современное понимание патогенеза GLD
На Рис. 2 изображена схема основного патогенетического механизма глобоидно-клеточной лейкодистрофии.
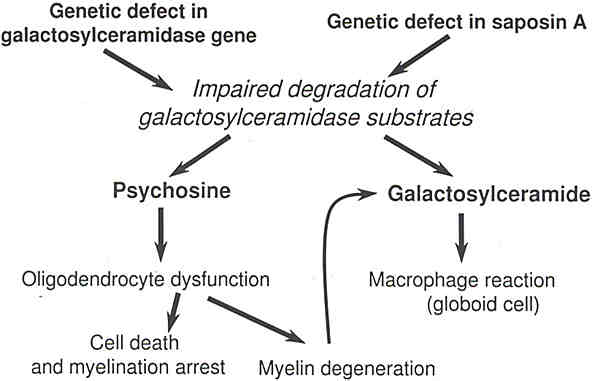 Рис. 2.Pathogenetic mechanisms operating in Krabbe disease as understood at this time.
Рис. 2.Pathogenetic mechanisms operating in Krabbe disease as understood at this time.
Важным дополнением к этой схеме является saposin A. Saposin A-дефицитная GLD в настоящее время известна только у экспериментальных мышей, но не исключено, что будут выявлены и такие больные. Таким образом, главными причинами заболевания могут быть генетические дефекты либо галактозилцерамидазы, либо saposin A. Их общим следствием является нарушенная деградация субстратов галактозилцерамидазы. А это означает, что в мозге накапливается галактозилцерамид и психозин (галактозилсфингозин). Тесное взаимодействие двух патогенетических механизмов может объяснить наиболее важные фенотипические проявления при болезни Краббе. Поскольку синтез и галактоцерамида и психозина ограничивается активно миелинизирующимися клетками, то процесс болезни не начинается до активного периода миелинизации. Это было четко продемонстрировано на мышах, имеющих «двойной» дефицит – нарушенный синтез галактозилцерамид/психозина и галактозилцерамидазы. Как только начинается миелинизация, начинаются и их метаболические превращения. Это приводит к появлению свободного галактозилцерамида в мозге больных из-за недостаточной деградации галактозилцерамида, который, в свою очередь, вызывает характерную глобоидно-клеточную реакцию. Галактозилцерамид синтаза также синтезирует психозин в активно миелинизирующихся клетках. В норме он немедленно распадается и никогда не превышает определенного уровня. При болезни Краббе аномальное накопление психозина достигает токсического для клеточного метаболизма уровня. Это ведет к быстрому и почти полному исчезновению олигодендроцитов. Психозин является также потенциальным индуктором апоптоза, как и С6 церамид. Клеточная гибель ведет к дальнейшему разрушению уже сформированного миелина, что способствует накоплению галактозилцерамида. С другой стороны, миелинизация прекращается на очень ранних стадиях развития из-за почти полной утраты олигодендроцитов. Этим можно объяснить парадоксальные характерные особенности заболевания, при котором первичный субстрат дефектного фермента не накапливается в аномально высоких количествах в ткани мозга.
Saposin A-дефицитные мыши показали, что несмотря на то, что два патогенетических механизма тесно взаимодействуют между собой, до некоторой степени и при определенных условиях они могут работать и автономно. Аномальное накопление психозина у saposin A-дефицитных мышей только в два раза выше нормы. Это не соответствует тому, что было найдено у больных, twitcher мышей и у других модельных животных при недостаточности галактозилцерамидазы, когда накопление психозина достигало 20-30 кратных значений по сравнению с нормой. По данным исследований
in vitro маловероятно, что двукратный уровень психозина достаточен для индукции цитотоксического эффекта на клетки в процессе миелинизации и апоптоза. Таким образом, у saposin A-дефицитных мышей заболевание может быть в первую очередь обусловлено глобоидно-клеточной инфильтрацией и связанной с ней иммунно-воспалительными явлениями, а не цитотоксичностью психозина. В таких случаях олигодендроциты выживают намного дольше, чем у мышей twitcher и деструкция уже синтезированного миелина будет протекать медленнее. Это также снижает скорость глобоидно-клеточной инфильтрации. Вполне возможно, что сходная автономная работа этих механизмов может происходить при медленно прогрессирующей форме GLD с поздним началом.
Будущие исследования
Автор полагает, что, несмотря на то, что основной патогенетический механизм болезни Краббе достаточно хорошо изучен, все еще остаются неясные моменты. Очевидно, что иммунные и воспалительные процессы играют важную роль в патогенезе заболевания, однако точный механизм их участия еще не определен. Интересными представляются наблюдения того, что как галактозил, так и глюкозил-психозин (glucosyl-psychosine) могут вызывать в тканевой культуре появление многоядерных клеток, напоминающих глобоидные клетки, и возможно психозиновые рецепторы. Эти находки могут значительно расширить наши представления о процессах, происходящих при GLD. Оба вещества галактозил- и глюкозилпсихозин в равной степени способны генерировать многоядерные клетки, но никогда не вызывают появление таких клеток in vivo при имплантации в мозг. Они крайне цитотоксичны, вызывают массивный геморрагический инфаркт и гибель экспериментальных животных. Известно, что только галактозилцерамид индуцирует глобоидно-клеточную реакцию при его имплантации в мозг. Известно также, что глюкозил-психозин накапливается в мозге больных с нейропатологической формой болезни Gaucher. Психозиновый рецептор имеет сходные константы с галактозилом и глюкозил-психозином. Высокая константа связывания рецептора может указывать на то, что природными лигандами(ом) для этого рецептора может быть не психозин. Несмотря на эти противоречия, исследования должны быть продолжены, поскольку они могут дать важную информацию для понимания патогенеза глобоидно-клеточной дистрофии.
Сайт создан в системе
uCoz