Авт. выявили у гетерозигот существенное снижение количества моторных нейронов в передних рогах спинного мозга мутантных мышей по сравнению с контролем. Достоверная нейрональная потеря выявляется у старых мышей (16 мес. у
и 19 мес. у Loa), у гомозигот большинство этих нейронов отсутствовало уже на 18.5 день эмбриогенеза (непосредственно перед рождением). Большинство из оставшихся нейронов подвергалось апоптозу, они обнаруживали Lewy-подобные включения, которые напоминали подобные тельца при ALS у человека.
Ни
Loa, ни
Cra1 мутации, по-видимому, не затрагивают существенно DHCHC белок, судя по уровню его экспрессии или субклеточной локализации. Поэтому авт. анализировали влияние мутаций на некоторые специфические функции DNCHC1. Ядерная подвижность во время клеточных делений и морфология и локализация Гольджи оказались неизменными. Однако, если аппарат Гольджи был нарушен с помощью агента, деполимеризующего микротрубочки, nocodazole, то его сборка была существенно нарушена у гомозиготных мышей
Loa, это привело авт. к предположению, что мутация проявляет свой эффект только в условиях стресса.
Loa гомозиготные мыши обнаруживали также дефекты в миграции лицевых нервов во время развития. В результате аномальной иннервации лица новорожденных возможно не м. сосать, что обусловливает их перинатальную гибель.
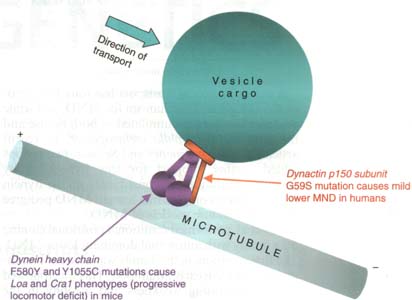
Т.о., цитоплазматический динеин наиболее важный белок для ретроградного транспорта в нейронах, то авт. анализировали мутантов
Loa в отношении аксонального транспорта. Выявлено снижение частоты высоко скоростных переносчиков в моторных нейронах спинного мозга с 67% в норме доя 21% у мутантов.
Итак, эти работы продемонстрировали прямую связь между дефектами аксонального транспорта и возникновением спонтанно появляющихся MND у людей и мышей (Рис. 1).
Сайт создан в системе
uCoz)