Мутации NPC1, идентифицированные у пациентов и исследования экспрессии мутантных кДНК пролили свет на взаимоотношения между структурой и функцией этого белка. Корреляции показывают, что три NPC1 домена являются критическими для функционирования: SSD, большая просветная богатая цистеином петля и просветный домен NPC1. Все мутации, локализованные в SSD, включая миссенс мутации, очень вредны, соответствуют гомозиготному состоянию отсутствия зрелого NPC1 белка, и имеют очень тяжелый биохимический и клинический фенотип (40,83). Изучение экспрессии SSD мутантов подтвердило критическую роль этого домена (84). Богатая цистеином просветная петля содержит приблизительно 50% всех описанных миссенс мутаций (включая 3 наиболее распространённые мутации), ассоциированных с изменчивой клинической и биохимической картиной. В гомо-аллельном состоянии I1061T коррелирует с нейрологической формой, начинающейся в юношеском возрасте, но сопровождается фатальной перинатальной б-нью печени. Пациенты, гетеро-аллельные по I1061T представлены всеми клиническими фенотипами, за исключением тяжёлой инфантильной нейрологической формы (20,40). Мутации, ассоциированные с биохимическим вариантом ( умеренные нарушения переноса клеточного холестерола) локализуются в петле, богатой цистеином, за 2 исключениями (40,73,74,80,81) (Рис. 3). Обзор опубликованных данных показывает образование кластера внутри небольшой области этой петли (Рис. 3). Гомозиготы по Р1007А и V950M соответствуют начинающейся у взрослых нейрологической форме. Гомозиготность по G992R описана у 63-летней женщины с не-нейрологической формой. Присутствие одиночного варианта этого аллеля, по-видимому, достаточно для обеспечения отличающегося биохимического фенотипа, который часто ассоциирует с медленно прогрессирующей начинающейся у юношей или взрослых формой, хотя это не всегда так (40,85). С др. стороны, классический биохим. фенотип постоянно наблюдается при более тяжелых нейрологических формах, но м. также ассоциировать с любой клинической формой болезни, включая взрослую форму без нейрологических проявлений. Мутации NPC1 домена (C63R, Q92R, C113R и Т137М), по-видимому, коррелируют с тяжелой инфантильной нейрологической формой (74,79,80,86).. Избыточно экспрессирующийся C113R белок не локализуется в поздних эндосомах, а скорее в эндоплазматическом ретикулёме, Rab7-негативных эндосомах и на клеточной поверхности (86). Мутации, затрагивающие или законсервированные остатки цистеина или мотив лейциновой застёжки в этом домене
дают неактивный белок,, направляемый в лизосомные мембраны, окружающие холестеролом-нагруженные сердцевины (84,87).
NPC1 polymorphisms
Описано более 50 экзонных и интронных SNPs (70,71,80-83). Наиболее превалирующими полиморфизмами являются Y129Y, H215R, P237S, I642M, I858V, N931N и R1266Q. Неожиданно I642M был описан в качестве мутации, вызвавшей болезнь у одного пациента (78). P237S SNP впервые описан как вредная мутация (40,83), и лишь позднее переклассифицирован в полиморфизм (68). В финской и шведской популяции 5% аллелей несут замены P237S, а некоторые NPC1 пациенты описаны, как имеющие две мутации в дополнение к P237S замене (74,86). Избыточная экспрессия P237S определенно указывает на то, что P237S является доброкачественным полиморфизмом, а не повреждающей мутацией (86).
NPC2 gene and protein
Используя протеомный подход, ген
НЕ1 картирован в хромосоме 14q24.3, который кодирует белок, первоначально описанный как главный секреторный белок человеческого epididymis, он оказался мутантным у NPC2 пациентов (15) и был переименован в NPC2. Он длиной в 13.5 т.п.н. и состоит из 5 экзонов. Зрелый в 132 аминокислоты NPC2 генный продукт является растворимым гликопротеином, экспрессирующимся повсеместно во всех изученных тканях (15). NPC2 связывается с манноза-6-фосфатными рецепторами и мигрирует совместно с лизосомными маркёрами в сахарозном градиенте (15). У нормальных фибробластов иммунореактивность NPC2 обнаруживается в транс-Гольджи сети, Lamp-1-позитивных эндосомах и Lamp-1-негативныйх периферических органеллах (86). Предполагается потенциальная роль NPC1 в качестве регулятора транспорта NPC2 (86). НЕ1 гомолог свиней соединяется с холестерлом с макромолекулярным сродством (88). Недавно получены доказательства (89) для значительно более высокого сродства связывания и идентифицирован гидрофобный связывающий холестерол карман около аминокислоты К97 (соответствует К116, если считать от инициирующего кодона). В независимом исследовании с кристаллической структурой высокого разрешения телячьего NPC2 эти данные подтверждены и выявлена рыхло упакованная область в белке ниже предполагаемого incipient холестерол-связывающего сайта (90). NPC2 обнаруживает 63% гомологию с Der p 2 dust-mite аллергеном. Хотя кристаллическая структура NPC2 и Der p 2 обнаруживает важные отличия, эти белки определенно составляют семейство для связывания гидрофобных лигандов. Предполагается, что NPC2 нуждается в конформационном изменении для связывания холестерола (90). Три области NPC2 белка считаются важными для функционирования, включая один для эффективной секреции (89).
Всего 11 семей известно с мутациями NPC2, все они обнаруживают заметные аномалии процессинга клеточного холестерола. Ни одна из известных на сегодня мутаций
NPC2 не затрагивает какой-либо из 4-х законсервированных доменов, но молекулярные исследования показывают высокие генотип/фенотипические корреляции (15-170. Большинство описанных NPC2 пациентов имеют начинающуюся после рождения тяжелую (часто летальную) респираторную или печеночную форму и/или тяжелую нейрологическую болезнь с гибелью до 4-летнего возраста. Почти все они обнаруживают нонсенс или сдвига рамки мутации, а Е20Х отвечает почти за половину мутантных аллелей. Две сестры с начинающейся в юношестве нейрологической формой и медленным течением имели splice-мутацию, ведущую к множественным транскриптам и менее тяжелой дисфункции белка (16). Два др. родственника с медленно прогрессирующим течением и начинающейся во взрослом возрасте нейрологической формой имели миссенс мутации (17).
Putative function(s) of the NPC1 and NPC2 proteins
Точные функции этих белков не установлены (6,8,19,91). NPC1 компартмент выглядит динамической, стерол-модулирующей сортинг-органелой. NPC1 белок в живых фибробластах показывает, что этот компартмент подвергается быстрым движениям, которые заметно нарушены в NPC1-мутантных клетках. Процесс, участвующий в продукции тубуло-везикулярных структур, который обнаруживает потерю гибкости и медленную скорость движения в мутантных клетках (92,93). Хотя точная схема остаётся неясной и некоторые результаты противоречивы, в целом же всё указывает на роль NPC1 в регуляции или осуществлении ретроградного транспорта множественных лизосомных грузов по позднему эндосомному/лизосомному пути (6,77). Помимо холестерола, гликолипиды также являются кандидатами для транспорта, зависимого от NPC1 (94,95), особенно если учесть. что они вместе с холестеролом и кавеолином, являются компонентами плотиков (raft). NPC1 компартмент обогащён гликолипидами, а интернализация GM2 ганглиозида в эндоцитотические пузырьки, как было установлено, нуждается в функциональном NPC1 белке (95). Показано, что транспорт эндосомы→Гольджи сфинголипидов м.б. блокирован с помощью высоких уровней внутриклеточного холестерола (96). Гомология NPC1 с прокариотическими пермеазами из resistence-nodulation-division (RND) семейства указывает на то, что белок NPC1 является трансмембранным молекулярным насосом, который м. действовать как пермеаза-транспортирующая жирные кислоты через клеточную мембрану (91). Как это согласуется с ролью NPC1 в клеточном перераспределении эндоцитозированного холестерола и гликолипидов, пока неясно (19). Регуляция транспорта субклеточных липидов м.б. фактически более сложной. Имеются строгие доказательства того, что NPC1 и NPC2 белки м. функционировать в тесном взаимодействии, т.к. их мутации не обнаруживают качественных отличий в их способности отвечать на нагрузку экзогенного LDL холестерола и на его тканевые липидные хранилища (12). Роль и NPC1 и NPC2 в регуляции гомеостаза стеролов, как полагают, в генерации из LDL холестерола продуцируемых oxystyerols (55). Недавно (89) подтверждено, что NPC2 специфически соединяется с холестеролом с высоким сродством. Некоторые белки, располагающиеся в эндосомно-лизосомной системе, NPC1, NPC2, MLN 64 и MENTHO (97,98) м. последовательно или альтернативно вовлекаться в перемещения клеточного холестерола (19). Постулируется (19), что активность NPC1 v/ зависеть от предварительного действия NPC2, чтобы ввести стерол в эндосомные/лизосомные мембраны.
Хотя точная причина, вызывающая нейродегенеративный синдром при NPC остаётся неизвестной, но хорошо известно, что дефекты при NPC1 или NPC2 вызывают 'traffic jam' липидов в поздние эндосомы (58). Было показано (99), что внутриклеточный перенос холестерола и гликолипидов в NPC фибробластах м.б. драматически улучшен с помощью избыточной экспрессии Rab7 или Rab9/ Сходные результаты получены и в др. исследовании (100) с Rab9. Хотя эти данные вызывают ряд нерешенных вопросов (101) они м. указывать на новые пути терапии.
Diagnosis
Диагностика NPC легка у пациентов с типичными симптомами, такими как комбинированная спленомагалия, атаксия и вертикальные gaze параличи. Но имеются очень разные клинические проявления, особенно у детей и новорожденных, а нейрологическое начало м.б. задержанным вплоть до созревания или взрослого периода. NPC часто не обнаруживаются в отсутствие органомегалии, нечастой ситуации у детей позднего возраста и взрослых. Нет скрининг-теста с мочой и кровью для диагноза NPC, хотя активность chitotriosidase обычно повышена в 10-20 раз. Активность кислой sphingomyelinase всегада нормальная в лейкоцитах (что позволяет исключить Nieman-Pick И или А) и иногда частично дефицитна в культивируемых фибробластах. Обычно присутсвуют foam клетки и sea-blue histiocyte в костном мозге, но м. и отсутствовать. Если они присутствуют, то foam клетки окрашиваются позитивно folipin. Ультраструктурные исследования биоптатов конюктивы, кожи или печени м. дать строгое подтверждение диагноза, но при биопсии печени возможны ложно-отрицательные результаты (26). Анализ липидов в печеночных биоптатах является не специфическим (102). Картины магнитного резонанса и компьютерной томографии м.б. нормальными или обнаруживать атрофию мозжечка или коры или при тяжелых нейрологических инфантильных формах, изменения белого вещества мозга.
Выявление аномального процессинга клеточного холестерола важно для специфической диагностики NPC1 или NPC2. Процедура нуждается в живых культивируемых клетках, обычно в фибробластах кожи. Убедительная диагностика м.б. достигнута при демонстрации накопления лизосомами неэфиризированного холестерола по интенсивной околядерной флюоресценции после окрашивания filipin (polyene антибиотика, который связывает гидроксильную группу неэфиризированного холестерола, чтобы сформировать флюоресцентный комплекс (Рис. 2) в сочетании с изучением внутриклеточного гомеостаза холестерола, определяемого по ранней скорости (4-6 ч) образования LDL-индуцированного choledteryl ester (66). В обоих тестах клетки сначала культивируются в среде, дефицитной по липопротеину, а затем помещаются в среду обогащённую LDL. Filipin тест, по-видимому, более специфичен (нарушение эфиризации м. происходить и при др. нарушениях) и более чувствителен. Большинство пациентов (80-85%) обнаруживают выраженные аномалии при в filipin тесте и в тесте на эфиризацию (классический фенотип). Остальные (отклоняющиеся фенотипы) обнаруживают умеренное накопление в тесте на filipin при скорости эфиризации от 30 до 80% от номы. Диагностика в этиз случаях трудна. Фибробласты при I-клеточной болезни ведут себя очень сходно с клетками NPC (66).
Сходные тесты м.б. использованы для пренатального обнаружения затронутых плодов, используется обычно культивируемые клетки хориональных ворсинок и клетки амниотической жидкости (103). Если мутация идентифицирована у пробанда, то пренатальная диагностика преимущественно достигается использованием молекулярно генетического тестирования. Некультивируемы хориональные ворсинки позволяют дать быстрый ответ в том числе в семьях с неклассическими формами (104). Свыше 200 беременностей с риском NPC было исследовано по всему миру, в большинстве случаев с помощью обычных биохимических методов. Идентификация мутаций у пробандов позволяла точное генотипирование у его родственников.
Treatment
Не существует специфического лечения для NPC. Трансплантации печени корректируют дисфункцию печени, но не влияют на прогрессирование нейролоической болезни. Определенный успех, достигнутый с помощью трансплантаций печни у
npcnih модельных мышей (105), неубедителен. Не выявлено одним из авторов долговременного благоприятного эффекта этой процедуры у пациентов . Сходным образом, трансплантации костного мозга у людей (106) у у животных моделей2 не оказывали существенного влияния на нейрологическое прогрессирование б-ни.
Фармакологические испытания были направлены на 2 мишени: снижение клеточного притока холестерола или снижение притока менее сложных гликолипидов. Изучение диеты с низким содержанием холестерола и варьирующими комбинациями холестерол-снижающих агентов (lovastatin, niacin и cholesteylamine) вызывают заметное снижение накопления в печени неэфиризированного холестерола (107), но не выявляется чётких доказательств успешности терапии при длительном использовании (6,29). Не выявлено успеха подобных диет и у модельных животных (108,109). В связи с дисфункцией головного мозга казалось рациональным использование substrate balance терапии с применением ингибиторов glucosylceramide synthase. У
npcnih мышей это не приводило к улучшению (51). С др. стороны, оральное применение N-butyldeoxynojirimycin у
npcnih мышей приводило к 20% задержке начала симптомов и увеличению выживаемости (110). Испытания с терапией с помощью N-butyldeoxynojirimycin (OGT 918) проводились для лечение пациентов с типом I Gaucher б-ни (111). Это лекарство (Miglustat, Zavesa) недавно было одобрено Европейским сообществом, Израилем и США. Клинические испытания этого агента проводятся сейчас и с NPC. В др. терапевтическом испытании на мышах использование neurosteroid ALLO, 3α 5α tetrahydroprogesterone задерживало начало и прогрессирование нейрологических симптомов и увеличивало количество клеток Пуркинье (112).
Подобной терапии на людах не проводили. NPC2 белок является растворимым и соединяется с манноза-6-фосфатными рецппторами и его добавление в культуральную среду клеток вызывало корректное клеточное накопление холестерола (15). Стратегии, разрабатываемые для дефицитов по лизосомным энзимам, следовательно, м.б. приложимы и к данному заболеванию. Дополнительные проблемы возникают с NPC1 белком, т.к. он, по-видимому, не секретируется и не замещается. Недавно избирательная избыточная экспрессия NPC1 трансгена в головном мозге npcnih мышей показала устранение как нейродегенерации, так и нейрологической симптоматики у трансгенных животных (113). Исследование подтвердило независимость нейральной и висцеральной патологии и показало, что избыточная экспрессия гена хорошо переносится.
Сайт создан в системе
uCoz 
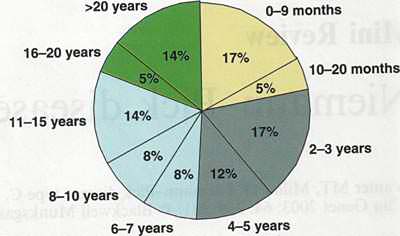 Возрастное распределение пациентов с болезнью Нимана-Пика типа С на момент диагноза. Данные по 200 пациентам.
Возрастное распределение пациентов с болезнью Нимана-Пика типа С на момент диагноза. Данные по 200 пациентам.
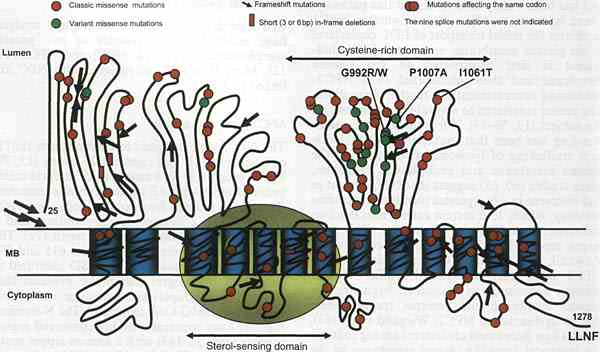 Топология мутаций NPC1. Схематическая модель белка NPC1 по Davies and Ioannou (72).
Топология мутаций NPC1. Схематическая модель белка NPC1 по Davies and Ioannou (72).