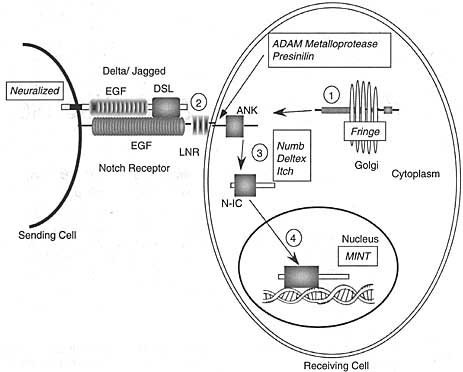
Во время ранних стадий развития сосудов предшественники эндотелиальных клеток дифференцируются и сливаются в сеть гомогенного размера примитивные кровеносные сосуды в процессе васкулогенеза. Более позднее развитие связано со сложным процессом ремодлирования и очистки, с пролиферацией, ветвлением и удалением лишних из вновь образованных сосудов в процессе ангиогенеза. N1 и N4 экспрессируются в эндотелиальных клетках в эмбриональной васкулатуре и, по-видимому, обладают частичной функциональной перекрываемостью. В то время как N4-/- мыши являются нормальными, N1-/- или N1/N4 двойные мутантные эмбрионы обнаруживают тяжёлые дефекты ангиогенного васкулярного ремоделирования (Табл.1) (29,38). Сходный васкулярный фенотип наблюдается и когда N4
экспрессируется у эмбрионов мыши (64), указывая тем самым, что соотв. уровни передачи сигналов Notch являются критическими для собственно развития эмбриональных сосудов. Notch лиганд Dll-1 (62) и Jag-1 (65) регулируют передачу сигналов Notch в этом контексте, т.к. неспособность экспрессировать эти лиганды во время эмбриогенеза мыши вызывает гемморагии и дефекты ремоделирования сосудов.
Первый ген Notch у млекопитающх был открыт, когда субнабор Т клеток при острой лимфобластной лейкемии, как было установлено, связан с конституитивной экспрессией N1
IC белка (66). Последующие исследования показали, что эктопическая активация N1 в предшественниках Т клеток вызывает их малигнизацию с высоким показателем и коротким латентным периодом (50, 67-70). Избыточная экспрессия N3
IC индуцирует Т-клеточную лейкемию, сходную с той, что индуцируется N1
IC, но физизиологическая функция N3
IC в развитии Т клеток неизвестна. Эти данные указывают на то, что нормальная функция N1 в регуляции пролиферации и дифференцировки Т-клеточных предшественников д. тонко контролироваться, т.к. передача сигналов Notch м.б. легко свёрнута, что способствует Т-клеточному онкогенезу. Сходным образом, функции N2 в созревании В клеток в селезенке м.б. ко-опитированы в некоторых случаях с В-клеточной хронической лимфоцитарной лейкемией (71). Интересно, что белок EBNA2, кодируемый Epstein-Barr virus (EBV, по-видимому, воспроизводит передачу сигналов Notch путём взаимодействия с CSL, индуцируя экспрессию Notch-регулируемых генов в отсутствие активации Notch (71, 72). Этот механизм скорее всего отвечает за способность EBV индуцировать В клетки и др. лимфомы.
еханизмы, с помощью которых N1 активация мндуцирует Т-клетоную лейкемию не совсем ясен, недавние данные указывают на то, что Notch м. кооперироваться с др. онкогенами в этой связи. Girard et al., использовали провирусных инсерционный мутагенез для идентификации генов, которые действуют вместе с
с-myc по ускорению развития CD4/CD8 Т-клеточных лимфом у мышей и нашли 52% конституитивно экспрессируемых активных версий N1. N3
IC-индуцированных Т-клеточных лимфом экспрессируют постоянно активный nuclear factor kappa B (NF-κB), регулятор клеточной жизнеспособности, пролиферации и продукции цитокина (73, 74). Анализ DN субнабора у N3
IC трансгенных мышей выявил накопление поздних DN тимоцитов, для которых характерны анти-апоптические эффекты передачи сигналов NF-κB в ответ на передачу сигналов пре-TCR. Интересно, что N1
IC непосредственно взаимодействует с NF-κB посредством ankyrin повторов
in vitro, способствуя трансактивации NF-κB в низких концентрациях, т.к. при высоких концентрациях отмечается ингибирующий эффект (75). Следовательно, N3
IC м. взаимодействовать с NF-κB посредством того же самого механизма. Недавно Beverly и Capobianco было показано, что доминантно-негативные изоформы Ikaros, лимфоид-ограниченного транскрипционнного фактора, увеличивают показатели и укорачивают латентный период Т-клеточной лейкемии у N1
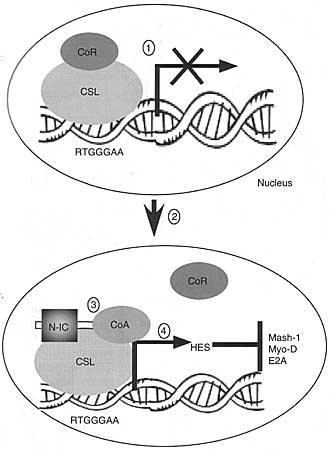
NC трансгенных мышей. Ikaros функционирует как транскрипционный репрессор во многих контекстах и интересно, что Ikaros и CSL соединяются с общей стержневой последовательностью, TGGGAA (76? 77). Поэтому было предположено, что, что во время N1
IC-индуцированного Т-клеточного лейкемогенеза, гены, которые обычно репрессируются с помощью Ikaros, экстопически активируются с помощью Notch. Следовательно, подтвержается, что Notch-индуцированный Т-клеточный онкогенез зависит от дисрегуляции комплекса, взаимодействующего с сигнальными сетями.
Дисрегуляция передачи сигналов Notch м. также вызывать неопластические трансформации в др. типах клеток, чьё развитие регулируется Notch. Напр., передача сигналов Notch регулирует способность панкреатических клеток предшественников выбирать между эндокринным и экзокринным клеточным клоном (78-80), а недавние результаты подтвердили, что эктопическая активация Notch вызывает экспансию метапластического эпителия протоков, первичное повреждение при раке поджелудочной железы у человека (81). Последующие исследования показали, что передача сигналов Notch действует иерархически ниже др. фактороd роста, таких как TGF-α и EGF, при индукции опухолей поджелудочной железы. Онкопротеин Ras м.б. ключевым промежуточным фактором между передачей сигналов ростовых факторов и активацией Notch, т.к. передача сигналов М является существенным нижестоящим эффектором онкогенного Ras и является существенной для поддержания неопластического фенотипа Ras-трансформированных клеток (82). Дисрегуляция функции белков, которые модифицируют передачу сигналов Notch также м.б. онкогенной, на это указывают недавние находки, что хромосомные транслокации, изменяющие функцию Notch коактиватора, Mastermind-like 2 (MAML2), превалируют в мукоэпидермоидных карциномах, наиболее распространённых злокачественных опухолей слюнных желез у людей (83).
В дополнение к способности аберрантной активации Notch способствовать неопластической трансформации многих типов клеток, недавно было показано, что N1 действительно функционирует как опухолевый супрессор в коже. Nicolas et al., показали? что условные делеции N1 в коже вызывают гиперпролиферацию базального эпидермального слоя, кульминацией чего является развитие кожных опухолей в 95%. Боле того, N1 нехватки облегчают развитие химически индуцируемых кожных карцином (84). Пути передачи сигналов Notch м. осуществлять свой супрессирующий опухоли эффект в коже путём ингибирования пути передачи сигналов Wnt. Полученные данные подтверждают, что нормальной функцией Notch в регуляции само-обновления и выбора клеточной судьбы во время органогенеза, м.б. кооперативаня или антогонистическая роль в необластической трансформации в широком круге типов клеток.
Notch signalling and human disease
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalophathy (CADASIL)
CADASIL является аутосомно доминантным нарушением, затрагивающим главным образом артерии головного мозга, вызывая субкортикальную гибель ткани белого вещества. Оно ассоциирует с мутациями N3 и вызывает в результате деструкции vascular smooth muscle cells (VSMC) артериол, которые необходимы для собственно образования многослойных стенок сосудов в артериях (85). Наиболее заметным признаком CADASIL является позднее начало болезни, в среднем в возрасте 45 лет (86). Симптомы CADASIL включают субкортикальные ишемические инсульты, нарушения настроения, мигрени и наиболее заметна прогрессивная деменция и гибель спустя 15-20 лет после клинического начала. Лежащей в основе причиной болезни является системная артериопатия, благодаря прогрессивной потере VSMC и замещения их granular eosinophilic material (GOM) неизвестного состава вокруг базальной мембраны и внеклеточного матрикса VSMC (87-89).
Участие N3 в CADASIL впервые было продемонстрировано с помощью позиционного клонирования в одной из семей, позволившего картировать эффект хромосомы 19 (90). Анализ сцепления др. семей с историей CADASIL позволил сузить искомую область до 800 т.п.н. (91), в которой был идентифицирован мутантный N3 ген. CADASIL характеризуется накоплением внеклеточной области N3 (N3
EC) на клеточной мембране вблизи GOM (92). Накопление N3
EC в васкулатуре кожи используется для диагностики CADASIL (93). Однако др. доказательства указывают на то, что N3
EC накопление и GOM м.б. вторичными следствиями нарушений закрепления VSMC во внеклеточном матриксе, вызываемых мутациями в N3 (44). Следовательно, механистические взаимоотношения между N3
EC мутацией и аккумуляцией, отложением GOM ? дегенерацией VSMC и симптомами CADASIL неясны.
Действительно все CADASIL N3 мутации участвуют в добавлении или потере цистеиновых остатков в одном из 34 EGF повторов внеклеточного домена (особенно в кластере первых 5 повторов, или за счёт непосредственно миссенс мутаций или мутацций сплайс-сайта) (94). Эти изменения ингибируют нормальное формирование трёх дисульфидных связей на EGF повтор, тем самым меняется структурная конформация белка. Это м. в свою очередь нарушать связывание лиганда, димеризацию, пост-трансляционные модификации, трафик клеток (95) или очистку из мембран (92) N3, каждое из них м. вызывать или усиление или уменьшение передачи сигналов. Усиление передачи сигналов N3 недавно было связано со снижением апоптоза в VSMC (96). Однако, при CADASIL не ясно, или происходит усиление сигнала N3, что вызывает накопление "поврежденных" VSMC или снижение сигнала N3 вызывает апоптоз во всём остальном здоровых клеток. VSMC существенны не только для формирования сосудистой архитектуры (85), но также высвобождают фактор проницаемости, vascular endothelial growth factor (VEGF), который увеличивает перфузию крови (97, 98). Снижение высвобождения VEGF м. снижать кровоток в определнных областях головного мозга, как это наблюдается у пациентов с CADASIL (99), это в свою очередь ведёт к инсульам и симптомам CADASIL. Выяснение функции N3 в VSMC (100) и роли мутантного N3 в качестве причины CADASIL проливает свет на структурно-функциональные взаимоотношения внутри N3.
Alagille syndrome
AGS в отличие от поздно начинающейся CADASIL является болезнью развития с клиническими признаками, обычно проявляющимися в течение первых двух лет. Это аутосомно доминантное, мультисистемное заболевание, характеризующееся дефектами печени, сердца, скелета и глаз, дающими в езультате широкий круг симптомов и величину смертности в 15-20% (101).Идентифицированы Jagged-1 мутации как ответственные за AGS (102, 103), по-крайней мере, у 70% пациентов (104). Цитогенетический делеционный анализ области 20р12 подтвердил, что AGS является результатом изменений в одиночном гене (105), идентифицированном как Jagged-1. Делеция всего гена Jagged-1 обнаруживается у 3-7% от всех AGS пациентов, указывая тем самым, что гаплонедостаточность является причиной, по крайней мере, некоторых случаев AGS. Из оставшихся известных мутаций Jagged-1 83% связаны с укорочением Jagged-1 из-за ненсенс мутаций или мутаций сплайс-сайта (106). Остальные 17% связаны с миссенс мутациями, образующими кластер вокрут внеклеточной части Jagged-1, кодирующей область DSL и первый повтор EGF (106). Тщательное исследование этих мутаций м. позволить выяснить нормальный механизм передачи сигналов или стабильности белка Jagged-1. Хорошо известные генотип-фенотипические взаимоотноешия у людей связаны с мутациями законсервированного глицина, это обусловливает аномальное гликозилирование субпопуляции Jagged-1
in vitro, не позволяя ей достигать клеточной поверхности. Пациенты с этой мутацией имеют 50-100% от нормального уровня Jagged-1? что приводит в результате к кардиальным дефектам, но не к аномалиям печени (107). Сильно варьирующая экспрессиия AGS у пациентов с одной и той же мутацией вместе с тем фактом. что 30% AGS пациентов не имеют известных Jagged-1 мутаций, указывает на то, что участвуют и др. средовые или генетические факторы, такие как вышестоящие или нижестоящие модификаторы и регуляторы Notch. Анализ сцепления у таких пациентов поможет выяснить др. критические части пути Notch в развитии у человека.
Spondylocostal dysostosis
Spondylocostal dysostosis (SD) является семейством родственных заболеваний, где дефекты поозвонков ассоциированы аномалиями рёбер и карликовостью из-за короткого туловища. Одна из специфических форм аутосомно рецессивного SD обусловливается мутациями в Dll-3 и характеризуется аномальной сегментацией остистых отростков, скорее всего из-за потери функции Dll-3 во время сомитогенеза (61) и в сегментационных часах, как было описано выше (108). Недавно 17 различных мутаций в Dll-3 было выявлено, которые связаны с укорочением белка, добавлением или делецифми цистеиновых остатков внутри EGF повторов или замещением существенных аминокислот (109). Открытие Dll-3 мутаций в субнаборе пациентов с SD открывает важность пути пердачи сигналов Notch в развитии скелета.
Сайт создан в системе
uCoz