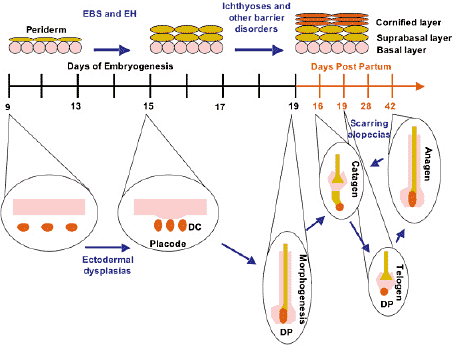
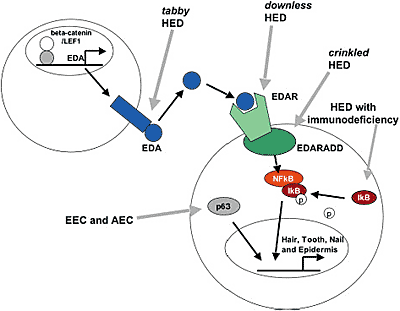
Классическая эктодермальная дисплазия, также как и нарушение с фрагильностью кожи и синдром Gorlin вносят существенный вклад в область эпидермального развития, т.к. они позволяют оценить последовательность сигнальных каскадов, которые продуцируют зрелые волосяные фолликулы. Так дальше всего вверх на этому пути являются Eda и Edar, мутации обоих генов ведут к эктодермальной дисплазии и они существенны для правильного формирования плакод. Следующей сигнальной системой является βcatenin
(из которой Plakophilin-1 является важной частью), она делает возможной дальнейшее развитие плакоды в зачаток волоса. Передача сигналов Shh необходима затем для морфогенеза волосяного фолликула после образования зачатка волоса.
Эпидермис между фолликулами м.б. подразделен на три основных компартмента. Базальный эпидермис, где осуществляется пролиферация, супрабазальный эпидермис, содержащий постмитотические кератиноциты, предназначенные дифференцироваться, и слущивающийся слой, где продуцируются дифференцированные чешуйки. Во время этой дифференцировки осуществляются тонко регулируемые программы генной экспрессии, одним из наиболее изученных примеров является изменение в экспрессии кератинов с базального на супрабазальные слои. Кератиноциты продуцируют большие количества кератина для промежуточных филамент, что придает кератиноциам их характерную крепость. У людей известные наследственные нарушения, которые меняют правильное формирование кератиновых филамент. Мутации в базальных keratins 5 и 14 ведут к аутосомно доминантному заболеванию epidermolysis bullosa simplex (Coulombe et al., 1991, MIM:131760), при котором эпидермис покрывается волдырями при минимальных травмах из-за дегенерации и разрыва базальных кератиноцитов. Мутации в супрабазальном кератине, keratin 10, вызывают заболевание, epidermolytic hyperkeratosis,
который характеризуется сходной дегенерацией и образованием волдырей, но супрабазальном слое эпидермиса (Rothnagel et al., 1992, MIM:
600648), Мутации ладонно-подошвенного кератина 9, дают тот же самый фенотип, но ограниченный ладонями и подошвами (MIM: 144200,
Rothnagel et al., 1995). Трансгенные мышиные модели, у которых экспрессируется человеческий мутантный кератин на том же самом уровне что и кератин дикого типа, и продуцируется фенотип, аналогичный тому, что наблюдается при наследственных заболеваниях (Fuchs et al., 1992; Cao et al ., 2001). Почти во всех случаях мутаций кератинов, мутации интерферируют с областью, где необходимы кератиновые мономеры для собственно сборки промежуточных филамент. Мутации в др. кератинах обусловливают заболевания, которые имеют больше общего с эктодермальной дисплазией, чем с эпидермолизом. Мутации супрабазальных кератинов 6 и 16 обусловливают нарушение, известное как pachyonychia congenita (PC1, MIM: 167200), при которой имеются тяжелые дефекты ногтей, также как и ладонно-подолвенный гиперкератоз, фолликулярный гиперкератоз и ротовые leukoplakia (McLean et
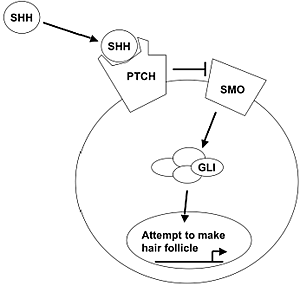 Рис. 3. The mammalian hedgehog pathway. Smoothened (SMO) can only signal to the protein complex containing Gli 1 when Sonic Hedgehog (SHH) is bound to Patched (PTCH). Under this circumstances, Gli 1 is translocated into the nucleus where it transcribes genes including PTCH which are responsible for the progression of the morphogenesis of the hair follicle, or when ectopically activated, causes the formation of a basal cell carcinoma.
Рис. 3. The mammalian hedgehog pathway. Smoothened (SMO) can only signal to the protein complex containing Gli 1 when Sonic Hedgehog (SHH) is bound to Patched (PTCH). Under this circumstances, Gli 1 is translocated into the nucleus where it transcribes genes including PTCH which are responsible for the progression of the morphogenesis of the hair follicle, or when ectopically activated, causes the formation of a basal cell carcinoma.
al ., 1995). Мутации в кератине 17 вызывают сходное нарушение (PC2,
MIM: 167210), но без лейкоплакий, но с присутствием зубов у новорожденных и кожных кист (McLean et al ., 1995). Чтобы еще больше усложнить вопрос и подчеркнуть гетерогенность, которая возникает у людей с кератиновыми заболеваниями мутации в keratin 17 м. также вызывать болезнь сальных кист, называемую steatocystoma multiplex (MIM: 184500). В некоторых случаях это два разных заболевания м.б. обусловлены одной и той же мутацией (Covello et al ., 1998). Мутации кератина 16 также м. вызывать focal non-epidermolytic palmoplantar keratoderma (MIM: 600962) (Shamsher et al., 1995). Конечной точкой эпидермальной дифференцировки является чешуйка, продукция которой тонко контролируется в гранулярном и слущивающемся слоях. Мутации в генах, необходимых для формирования чешуек, ведут к болезням недостаточности барьерной функции, известным как ichthyoses. Классический ихтиоз и первый для которого была описана мутация, был lamellar ichthyosis (MIM: 242300). При данном заболевании пациенты страдают от тяжелого кожного недуга, при котором образуются крупные чешуйки (отсюда название), ассоциированные с различной степенью покраснения. Болезнь обусловливается мутациями в гене transglutaminase (TG-1) кератиноцитов (Russell et al .,
1995). Этот ген существенен для образования чешуек, т.к. он делает возможным поперечное связывание различных поддерживающих белков слущивающейся оболочки с плазматической мембраной. Мыши, нокаутные по Tg-1
имеют сходный фенотип с покраснением, чешуйками и быстрым наступлением после рождения значительной транс-эпидермальной потери воды (trans-epidermal water loss (TEWL)) и гибели новорожденных (Matsuki et al ., 1998). Фактически имеются и др. нокаутные и трансгенные мыши, которые имеют TEWL , а затронутые гены почти всегда играют роль в поддержании барьерной функции. Естественно возникшая мутация у мышей, рецессивная
flaky tail, имеет фенотип, сходный с симптомами др. из таких болезней, ichthyosis vulgaris (MIM: 146700). Мыши имеют крупные чешуйки на подушках лап и хвосте, заметно пониженное количество клеток в гранулярном слое, а также акантоз и orthokeratotic hyperkeratosis, проникающие внутрь проекции эпидермиса с гиперпролиферацией эпидермиса, однако с нормальной экспрессией кератинов. Такие мыши имеют пониженные количества зрелого filaggrin и повышенные количества незрелого filaggrin, которые не способны подвергаться процессингу, чтобы продуцировать keratohyalin гранулы, важное составляющее при формировании чешуек (Presland et al ., 2000). Мыши
flaky tail кроме того обладают и др. признаками - сужениями хвоста и подушек конечностей - это является одной из основных характеристик др. аутосомно доминантного наследственного нарушения у людей, Vohwinkels syndrome,
обе классические формы характеризуются глухотой (MIM: 124500)
или variant form (MIM:604117). Vohwinkels syndrome является др. формой генерализованного ихтиоза. Часто обнаруживаются аутоампутации пальцев в результате перетязек в суставах пальцев рук и ног, называемых pseudoainhum. Мутации по cornified оболочечному белку loricrin ответственны за variant форму синдрома Vohwinkles (Maestrini et al., 1996), тогда как мутации connexin 26, кодирующего белок щелевых соединений, ответственны за классический синдром Vohwinkels (Maestrini et al ., 1999). Мутации в loricrin ассоциируют также в целом с идентичным заболеванием progressive
symmetric erythrokeratoderma (MIM:602036). Исследование с использованием трансгенных мышей, экспрессирующих эту мутантную форму loricrin, показало, что такие мыши имеют фенотип, очень сходный с обоими заболеваниями. Мутантный ген имеет сдвиг рамки считывания, что позволяет транслироваться др. 22 аминокислотам, которые, по-видимому, кодируют сигнал ядерной локализации. Мутантный loricrin накапливается в ядре и поэтому не откладывается в cornified оболочке (Suga et al., 2000). Спаривание нокаутных с трансгенными мышами вызывает усиление тяжести фенотипа. Мутантный
loricrin, по-видимому, оказывает доминантный негативнцй эффект, который является дозово-зависимым и предупреждает собственно образование безъядерных чешуек. Кератины также, по-видимому, играют роль в ichthyosiform заболеваниях. Как уже упоминалось, все др. генодерматозы с участием мутаций в кератинах обусловлены мутациями в начале и конце палочковидной области белка, важного собственно для сборки кератиновых филамент. Однако, новая мутация в вариабельной хвостовой области кератина 1, по-видимому, обусловливает тяжелый гиперкератоз, названный ichthyosis hystrix Curth-Macklin (MIM:146590). Эта мутация кератина нарушает способность loricrin транслоцироваться в десмосомы при подготовке к сборке cornified оболочек (Sprecher et al., 2001). Кератин 2e экспрессируется позднее в дифференцировке кератиноцитов. Спектр различных мутаций вызывает и др. ichthyosiform заболевание, ichthyosis bullosa of Siemens (Rothnagel et al., 1994, Kremer et al., 1994).
The desmosome and diseases of epidermal integrity
Эпидермис находится под воздействием значительных стрессов, как внешних, так и механических. Чтобы защитится от повреждений кератиноциты соединяются с помощью обширной сети межклеточных взаимодействий, называемых десмосомами. Заболевание ранимости кожи, обусловленное мутациями plakophilin-1, описанное выше, является болезнью, обусловленной аномалиями десмосом. Имеется несколько наследственных заболеваний, которые обусловлены мутациями в генах, кодирующих компоненты десмосом, которые обычно дают в результате фенотип фрагильности кожи. Striate palmoplantar keratoderma является заболеванием, при котором пациенты имеют hyperkeratotic полосы на ладонях и подошвах. Болезнь наследуется по аутосомно доминантному способу и является результатом гаплонедостаточности десмоплакина или десмосоного cadherin, desmoglein 1, возникающей в результате делеции N-терминальной части (Armstrong et al., 1999, Rickman et al ., 1999, Whittock et al., 1999). Напротив, когда ген desmoplakin обладает рецессивными мутациями или компаундной гетерозиготностью, то возникает keratoderma с wooly волосками, ассоциированная с кардиомиопатией, обычно известная как Naxos disease (MIM:601214). Пациенты с Naxos disease м. также иметь мутации в armadillo повторы содержащем (βcatenin подобном) гене plakoglobin (Norgett et al., 2000). По крайней мере, существует еще один локус для Naxos disease (Djabali et al., 2002). Как и в случае с некоторыми эктодермальными дисплазиями не совсем ясно, почему мутации в генах д. вызывать фенотипические изменения кожи. Двумя такими заболеваниями являются Hailey-Hailey (MIM:169600) и Darier disease (MIM:124200). Hailey-Hailey Disease представляет собой повторяющиеся разрывы пузырьков и bullae на шее, в пахах и подмышечных впадинах, тогда как пациенты с болезнью Darier имеют keratotic papules, ассоциированные с сальными областями. Оба имеют субклинические аномалии межклеточных взаимодействий кератиноцитов. Гистологические исследования выявляют acantholysis, который отделяет кератиноциты в супрабазальной области эпидермиса. Мутации в sarco/endoplasmic reticulum calcium pump ATP2A2 ответственны за болезнь Darier, тогда как мутации ATP2C1
ответственны заe Hailey-Hailey болезнь (Sudbrak et al., 2000; Sakuntabhai et al., 1999). Благодаря широкому разнообразию генных мутаций, вызывающих эпидермальные дефекты, становится ясным, что дифференцирующийся эпидермис является тонко контролируемой тканью, легкие пертурбации в которой м. вызывать тяжелые аномалии.
Diseases of hair cycling and hair shaft formation
Аномалии волосяных фолликулов являются областью, в которой знание мышиных мутаций превосходят число болезней у людей. Присутствие вибрисс и синхронизация волосяных циклов являются двумя основными причинами, которые делают легкой детекцию незначительных изменений фенотипа волосяных фолликулов. Существует множество наследственных болезней человека, которые попадают в две основные категории, дефекты формирования остей волос и дефекты циклического образования волос. Циклы образования новых волос это специализированный онтогенетический процесс, зависящий от продолжающихся взаимодействий между мезенхимной тканью, дермальными сосочками и соответ. эпителиальными клетками или т.наз. раздутыми (bulge) клетками волосяного фолликула или быстро пролиферирующими матриксными кератиноцитами. Рост остей волос является результатом пролиферации матрикса и является чрезвычайно организованной комбинацией процессов формирования паттерна и дифференцировки, которая ведет к формированию зрелых остей волос, состоящих из 6 концентрически расположенных цилиндров специально дифференцированных кератиноцитов. Ости волос состоят, по крайней мере, из 100 белков, мутации некоторых из них ведут к уродствам остей волос. В качестве примера таких болезней м. служить monilethrix (MIM:158000), при которой пациенты имеют
редкие видимые волосы из-за их ломкости. Микроскопический анализ волос выявляет характерные цепочки (beading) по длине ости, которые, по-видимому. вносят вклад в сравнительно легкую ломкость остей. Мутации двух волосяных кератиноцитов, кстати, участвуют в этом заболевании, волосяной кератин 6 волосяной кератин 1 (Winter et al., 1997a; Winter et al., 1997b). Оба кератина экспрессируются в кортексе клеток волосяных фолликулов и преимущественно вносят вклад в физическую крепость остей зрелых волос. Подобно др. кератинопатиям , monilethrix является доминантно наследуемым заболеванием, отражающим механизм действия мутаций кератинов на сборку промежуточных филамент.
Рецессивный nude фенотип у мышей характеризуется врожденным отсутствием волос, а также тяжелой иммунологической недостаточностью. Аналогичное заболевание у людей (MIM:601705) характеризуется идентичным фенотипом. Фактически волосяные фоликулы присутствуют, но ости волос аномальны и не способны пенетрировать на поверхность. Мутации в winged helix гене Whn (winged helix nude, теперь называется Foxn1) ответственны за мутантных мышей, а также за ассоциированное заболевание у людей (Brissette et al., 1996; Frank et al., 1999). Как мутации гена Whn вызывают болезнь, неизвестно, предполагается, что фенотип обусловлен проблемами с правильной экспрессией кератинов в дифференцирующихся остях волос. Это ведет к характерной конволюции верхней части ости волоса, в результате он теряет способность пробиваться на поверхность кожи. Имеется большое количество нарушений, которые ведут уродствам остей волос. Сюда входит Netherton syndrome (MIM:256500), аутосомно рецессивный ichthyosis, обладающий характерными нарушениями остей волос, известными как trichorrhexis invaginata (bamboo hair). Мутации в гене SPINK5, кодирующем серин протеазный ингибитор LEKTI, также выявлены при синдроме Netherton (Chavanas et al ., 2000), скорее всего подобно гену WHN, функция этого гена, по-видимому, связана с образованием остей волос. Мутации в гене транспортера меди ответственны за X-сцепленное заболевание Menkes kinky hair syndrome (MIM:309400, summarised by Davies, 1993), при котором пациенты имеют редкие волосы, а также неврологические нарушения. Микроскопический анализ выявляет pili torti (скрученные волосы), trichorrhexis
nodosa (фокальные переломы ости волос) и гипопигментированные волосы.
Снова биологическая функция гена транспортера меди для формирования нормальных остей волос неизвестна. Недавно выявлена связь между межклеточной адгезией и интегральностью остей волосв результате открытия гена, мутантного у мышей и крыс
lancolate и при соответ. болезни людей localised autosomal recessive hypotrichosis, LAH, который обусловливает продукцию крупных комочков на растущих остях волос, которые прерывают ость волоса, когда он прободает эпидермис и дает внешний вид "lance-head" (Kjulic et al., 2003; Jahoda et al., 2004). Во всех этих случаях мутации в новом десмосомном кадхерине, desmoglein 4
(Kjulic et al., 2004 and Whittock and Bower, 2003), вызывают тяжелые аномалии в межклеточной адгезии в волосяном фолликуле, а также средней степени гиперплазию межфолликулярного эпидермиса, по-видимому, он необходим для корректных межклеточных контактов для поддержания кожи и волос.
Как уже упоминалось выше после морфогенеза волосяной фолликул проходит через цикл из трех самостоятельных фаз. Сначала фаза активного роста или anagen, затем фаза инволюции, называемая catagen, которая затрагивает и дермальный сосочек, мезенхимный органзующий центр волосяного фолликула и, наконец, фаза молчания, известная как telogen. Нарушения в anagen д., безусловно, приводить к изменению длины волос. Это имеет место в случае мышей
angora, где 50% рост волос обусловлен 50% продолжительностью фазы anagen как результат мутации в гене
fibroblast growth factor 5 gene (Fgf5), которая задерживает начало catagen (Hebert et al., 1994). Как уже упоминалось ранее, мутации, затрагивающие anagen легко выявляются у мышей с их сихнонизироваными циклами волос. Однако, мутации
angora у людей трудно определимы, т.к. фаза anagen у людей очень длинна от 3 мес. до 6 лет. Очень редки синдромы с избыточным ростом волос или гипертрихозами у людей, молекулярные основы таких гипертрихозов неизвестны. Во время новых циклов образования волосяных фолликулов после активной фазы роста стартует программа кератинизации остей волос, которая в конечном итоге формирует club волоса, который закрепляется в волосяном фолликуле во время telogen. Имеются заболевания у людей, при которых club hairs легко выпадают, такие как loose anagen syndrome или telogen effuvium (Sinclair et al., 1999, Rebora et al., 1997). Генетические основы этих заболеваний неизвестны. Информацию получили от мышей, нулевых десмосомному кадхерину demoglein 3 (dsg3), антигену pemphigus vulgaris. В дополнение к фенотипу pemphigus vulgaris мыши обнаруживают раннюю потерю волос во время telogen (Koch et al ., 1998). У дикого типа и у людей telogen фолликулы обнаруживают интенсивное окрашивание антителами к dgs3 на поверхности кератиноцитов в контакте с club hair. Кстати, это первые доказательства гена, ответственного за закрепление волос во время telogen.
Заболевания, которые затрагивают способность волосяных фолликулов проходить через ст. catagen, обнаруживают сходный фенотип у мышей и у людей. Благодаря исследованиям мышей hairless (hr) было показано, что alopecia, ассоциированная с этим фенотипом является результатом дефектов в catagen (Panteleyev et al ., 1999). У мышей hairless повышена степень апоптоза в волосяных фолликулах в начале ст. catagen, что нарушает эпителиальный слой и это позволяет дермальным сосочкам возвращаться в выпячивание волосяного фолликула. В результате дермальные сосочки оказываются глубоко в дермисе, а волосяные фолликулы не способны продолжать цикл. Крупное отверстие в верхней части волосяного фолликула, называемое utricles, формируется и в некоторых случаях в них образуются крупные кисты. Фолликул никогда не м. снова приобрести способность в ступать в новый цикл и теряет способность продуцировать волос. Соотв. нарушение у людей известно (MIM:209500), оно затрагивает все волосы. Как и предполагалось мутации в гене
HR оказались ответственны за заболевание у огромного числа семей (Ahmed et al., 1998, Ahmed et al., 1999, Ahmed et al ., 1999,Aita et al ., 2000, Zlotogorski et al., 1998). Также родственная двум формам HED, имеется сходная болезнь, обусловленная мутациями в vitamin D receptor (VDR) у людей, а также у мышей, нулевых по
или VDR или retinoid X receptor β subunit (Miller et al .,
2001, Li et al ., 1997, Li et al., 2001).
Conclusions, future directions, complex traits
The exploding field of trangenic mice and more recently the
advent of tissue specific transgenics and knockout mice has
allowed the manipulation of not only genes implicated in human
monogenetic disorders (such as the keratins) but also the investigation
of genes that are normally critical for the development of
the organism. The combination of these approaches as well as
mouse mutants both identified and unidentified, has and will
continue to reveal large amounts of information on the development
and differentiation of the epidermis and its appendages, as
well as inherited disorders. There are still many mouse mutants
that have abnormalities in skin and hair as well as hair cycling.
Many of these mutants have no human counterpart to date, but
the hope is that important connections can be made between
mouse and man with the near completion of the sequence of both
genomes will continue to lead to the identification of new genes.
The use of comparative genomics approaches allows the linking
of mouse mutations to a syntenic region in the human and vice
versa.
Psoriasis and alopecia are at a are common diseases, the first
a disease of abnormal differentiation of the epidermis, the second
the reversible destruction of structures of the hair follicle. Both
diseases exhibit hallmarks of inheritance as complex genetic
traits (Aita et al., 1999, Barker 2000). The power of mouse models
has failed in trying to determine a basis of these diseases as they
are impossible to model in congenic mouse strains. Alternative
approaches must be used to zero in on the cause of diseases such
as these. Classical approaches such as linkage analysis and
analysis of affected pairs of siblings to discover shared alleles are
used to discover regions of the genome associated, linkage to
specific HLA alleles has also been investigated. However this is
still a long way from identifying the gene(s) responsible for either
psoriasis or alopecia areata. Coupling these approaches with the
comprehensive expression analysis afforded by microarray technology
will enable the detection of differentially expressed families
of genes in the regions of interest and has already been
perform to some degree in alopecia areata. In addition, widescale
screening of SNPs, single nucleotide polymorphisms, particularly
in the coding regions of genes could lead to an elucidation of the
genotype of individuals with the disease and maybe recapitulation
of the disease in the mouse.
To conclude, the last five years have been interesting times in
the discovery of the molecular basis of many inherited disorders.
The benefit that this has had to the understanding of the basic
biology of the skin and its appendages cannot be over-emphasised.
In the next five years, there will no doubt be an even greater body
of knowledge accrued on the subject, as the discovery of mutations
in monogenetic disorders, more accurate chromosomal
linkage and the discovery of susceptibility loci in complex disorders
continues.
| |
|

ПРИРОДА НЕ УСТАЕТ
ТВОРИТЬ ЧУДЕСА
Генетика — штука коварная. Периодически она откалывает неожиданные номера.
Такое произошло и с этими жизнерадостными мальчишками — сыновьями 26-летней Юаны Гомец из деревни Лорэто, что на границе Мексики
с США. Юана родила двух мальчиков, сплошь покрытых темным волосяным покровом. Сегодня Габриэлю 11 лет (слева), Виктору 7 лет. Ребята физически и духовно развиты выше среднего. Они регулярно посещают школу в родной деревне, несмотря на некоторые проблемы, которые возникают
порой вследствие их экзотической внешности. Дело в том, что жизнерадостные «волчата» своеобразная генетическая жертва фамильного рода. Еще в 1905 году у их прапрабабушки Марии Луизы Днац появилась на свет дочь с такими же
странными отклонениями. С тех пор в семействе был зарегистрирован
21 нормальный ребенок —14 девочек и 7 мальчиков. И вот через десятилетия Габриэль и Виктор вдруг «подхватили» генетическую эстафету своих далеких
предков; явление редкое, но оно случалось в разное время и в других
странах, в частности, в России и Германии.
|
Сайт создан в системе
uCoz