Diabetes: The importance of the liverGuy A. Rutter
Сurent Diology 2000, 10: R736-R738 (Перевод В.К. Викуловой) |
Резистентность к инсулину, признак инсулин независимого diabetes mellitus, характеризуется неспособностью тканей поглощать и запасать глюкозу в ответ на действие инсулина. Две недавние работы проясняют значение пути передачи сигналов инсулина для печени и его нарушение при диабете.
Преобладающей формой диабета mellitus, инсулин независимого диабета mellitus (NIDDM) или диабета типа 2 страдают приблизительно 4% популяции Западного мира, на их лечение уходит более 10% общего бюджета National Health Service в Англии. Заболевание характеризуется неспособностью чувствительных тканей к нормальному ответу на инсулин - "инсулиновой резистентностью". Эти ткани оказываются неспособными к поглощению и хранению глюкозы, что приводит к хроническому увеличению количества глюкозы в крови (" гипергликемии"). В мягко выраженных случаях увеличенная продукция инсулина &beta-клетками островков Лангерганса в поджелудочной железе, приводящая к "гиперинсулинемии", может компенсировать уменьшение чувствительности к гормону, но диабет возникает когда клетки неспособны отвечать на гормон [1]. Причины заболевания остаются слабо изученными, однако преобладание больных среди индивидов с избыточным весом (80% - диабетики типа 2) заставляет предполагать, что нарушение регуляции жирового метаболизма может быть важным фактором заболевания.
За усваивание глюкозы из крови у здоровых индивидов отвечают три ткани - печень, мышцы и жировая ткань. Все три становятся при NIDDM резистентными к инсулину, но относительное значение каждой из тканей неясно. Две новых работы [2,3] проливают свет на этот вопрос. Michael et al. [2] показал, что инактивация гена инсулинового рецептора особенно в печени приводит к возникновению диабетоподобных симптомов у мыши. Т.е. инсулин играет непосредственную роль в метаболизме печени. Используя других модельных животных, Shimomura et al. [3] обнаружил, что в развитие инсулиновой резистентности в печени вовлекается избирательная инактивация способности инсулина блокировать продукцию глюкозы в печени. С другой стороны способность гормона стимулировать синтез жирных кислот сохраняется. Таким образом, наряду с освещением важности печени как мишени для действия инсулина эти работы также дают новую информацию о средствах, с помощью которых рецепторы к инсулину посылают сигнал внутрь клетки.
Потеря печенью рецепторов к инсулину приводит к гипергликемии
Michatl et al. [2] использовал метод прицельного выбивания (knockout) гена, кодирующего рецептор к инсулину, специфический для печени - рецепторную тирозин киназу. Как описано ранее [4], для этого производят скрещивание мышей, у которых ген рецептора к инсулину дикого типа заменен соответствующим геном с примыкающими сайтами lox, чувствительными к рекомбиназе, с животными, экспрессирующими рекомбиназу "Cre" в специфической ткани, в данном случае в гепатоцитах. Это приводит к выбиванию гена печень-специфического рецептора к инсулину ("LIRKO") и сильно выраженной инсулиновой резистентности у молодых животных (возраст - 2 мес.). Появляющаяся в результате явная толерантность к глюкозе контрастирует с отсутствием эффекта инактивации гена рецептора к инсулину в мышцах ("MIRKO")[5]. Этот результат был неожиданным, принимая во внимание, что скелетные мышцы отвечают за потребление более, чем 70 % глюкозы, сопровождающееся карбогидратной переработкой[6], тогда как печень отвечает за усвоение большей части оставшегося количества глюкозы[7].
Новые результаты продемонстрировали, что в норме инсулиновый сигнал в печени важен для дальнейшей утилизации груза глюкозы. Прежде предполагали, что увеличенная продукция глюкозы печенью при голодании и диабетах может возникать только непрямым образом вследствие отсутствия инсулина. Тот факт, что иньекция инсулина не приводит к подавлению продукции глюкозы печенью у животных LIRKO, одновременно подавляя липолиз и освобождение глюконеогенных предшественников из мышц, указывает на то, что прямое действие инсулина на эти органы должно быть важным для животных дикого типа.
Животные LIRKO имеют необычно высокие уровни (10-20 кратно превышающие нормальный) циркулирующего в крови инсулина в результате увеличения массы &beta-клеток островков (приблизительно в 5 раз по сравнению с нормальным уровнем), гиперсекреции инсулина и уменьшения удаления инсулина печенью[2]. Последнее можно было ожидать, принимая во внимание, что для включения гепатоцитами процесса внутренней переработки и разрушения гормона необходим активный рецептор инсулина. Механизмы, лежащие в основе такой сильной гиперплазии островков, отмечаемой и у других грызунов, являющихся моделями диабета [9], неизвестны, но высокие уровни в крови инсулина или глюкозы, или некоторых факторов развития печеночного происхождения могут быть важны. Интересно, что животные LIRKO не страдают от недостатка секреции инсулина даже после года увеличенной продукции инсулина - у человека же сверхпродукция инсулина, как полагают, вызывает "истощение &b-клеток" и открытый диабет[10]. Неясно, однако, насколько близко модельные мыши LIRKO отражают человеческое состояние, при котором гиперплазия &beta-клеток может быть менее резко выражена и, таким образом, нагрузка на предсуществующую популяцию &beta-клеток больше.
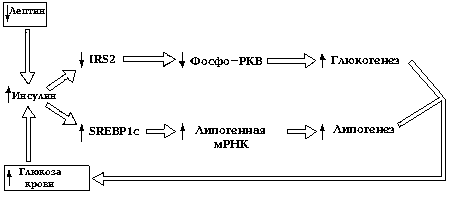
Рис. Схема, иллюстрирующая значения инсулинемии для возникновения резистентности к инсулину в печени.
Снижение уровня лептина (и увеличение уровня глюкозы в крови) провоцирует усиление выработки инсулина клетками панкреатических островков, приводящее к снижению уровня печеночного IRS2, к уменьшению активации PKB и к невозможности подавления глюконеогенеза. В то же время увеличения уровня SPREBP1c активирует гены, участвующие в липогенезе и освобождении жирных кислот. Жирные кислоты в сочетании с глюкозой стимулируют дальнейшее высвобождение инсулина.
Рис. Схема, иллюстрирующая значения инсулинемии для возникновения резистентности к инсулину в печени.
Снижение уровня лептина (и увеличение уровня глюкозы в крови) провоцирует усиление выработки инсулина клетками панкреатических островков, приводящее к снижению уровня печеночного IRS2, к уменьшению активации PKB и к невозможности подавления глюконеогенеза. В то же время увеличения уровня SPREBP1c активирует гены, участвующие в липогенезе и освобождении жирных кислот. Жирные кислоты в сочетании с глюкозой стимулируют дальнейшее высвобождение инсулина.
Избирательные изменения в передаче сигналов через печеночные инсулиновые рецепторы
Мыши LIRKO представляют эффективное новое средство для исследования важности инсулиновых сигналов для функции печени [2], однако дефекты рецепторов к инсулину сами по себе, как полагают, не играют значительной роли в патологии большинства форм NIDDM [11,12]. Michael et al. [2] использовал две модели резистенстности к инсулину для исследования экспрессии генов, способной объяснить возникновение нарушения в печени инсулинового сигнала. Первая модель - это хорошо известные мыши ob/ob, лишенные фактора насыщения, лептина (leptin), кодируемого геном obese(ob)[13]. Эти животные переедают, у них в избытке секретируется инсулин и возникает ожирение печени(fatty liver). Вторая модель была создана случайно при подавлении формирования жировой ткани - состояния, называемого липодистрофией - в результате экспрессии доминантно -негативной формы трнаскрипционного фактора, известного как фактор-1 дифференциации/ детерминации (ADD1) или как элемент связывания белка-1 при стерольном ответе (sterol response element binding protein-1 - SREBP1) [14]. В отсутствии отложений жира вне печени у этих мышей подобно животным ob/ob развивается ожирение печени (fatty liver) и возникает сильная резистентность к инсулину.
У животных обеих моделей резистентности к инсулину индуцирующее действие инсулина на экспресиию SREBP1 в печени [15] существенно не затронуто. Следовательно, SREBP1 способен трансактивировать липогенные гены, включая гены, кодирующие глюкокиназу, ацетил -СоА-карбоксилазу и синтетазу жирных кислот. Таким путем синтез липидов в печени, как и ожидалось, возрастает. В сравнении с этим, экспрессия генов, кодирующих ферменты глюконеогенеза, которые в нормальных животных подавляются инсулином [16] такие как фосфоэнол- пируват- карбокси киназа и глюкозо-6-фосфатаза, у животных-моделей остается на высоком уровне.
Почему инсулин не является эффективным в регуляции генов глюконеогенеза, но способен индуцировать работу генов липогенеза? Вероятное объяснение этого заключается в том, что ключевые молекулы в передаче сигнала от инсулинового рецептора к субстрату-2 инсулинового рецептора (insulin receptor substrate 2 - IRS2) были в обеих моделях сильно редуцированы [3]. IRS2 и другие изоформы IRS[12] в норме подвергаются фосфорилированию при связывании с инсулиновым рецептором тирозин-киназой, приводя к пополнению сигнальных молекул, особенно фосфатидилинозитол 3’-киназы. Это, в свою очередь, инициирует каскады белковых киназ - например, вовлекающих протеиновую киназу В, которые в конечном итоге реализуют действие гормона на транскрипцию и метаболизм [12]. В согласии с этими находками показано, что IRS2 является важным в возникновении сигнала от печеночных рецепторов к инсулину и менее важным для возникновения сигнала от рецепторов к инсулину в мышцах [18]. Отметим [3], что путь передачи сигнала от инсулинового рецептора к SREBPP1 в значительной степени не зависит от IRS2.
В сообщении Shimomura et al. [3] приводятся доказательства, что подавление функции гена IRS2 вероятно является результатом гиперинсулинемии. При культивировании гепатоцитов в присутствии повышенных концентраций инсулина мРНК гена IRS2 уменьшается, а нарушение &beta -клеток in vivo с помощью стрептозотоцина вызывало уменьшение экспрессии гена ISR2 в печени.
Направления дальнейших исследований
Эти результаты оставляют без ответа ряд интересных вопросов. Во-первых, вызывает ли постоянно повышенный уровень инсулина резистентность к нему в тканях, не относящихся к печени, и могут ли такого рода изменения быть объяснены снижением экспрессии IRS2? Неясно, влияют ли изменения в экспресси IRS2 в мышцах на резистентность к инсулину, так как в этой ткани большее значение имеет IRS1 [18].
Другой вопрос касается роли накопления липидов при снижении уровней экспрессии IRS2 в печени мышей ob/ob и мышей с липодистрофией [3]. Липотоксичность имеет отношение к дисфункции &beta -клеток [19] и может происходить отчасти вследствие сверхэкспрессии SREBP1 [20](C. Zhao and G.A.R., неопубликованные данные). В &beta-клетках поджелудочной железы функция IRS2 особенно важна для формирования подпитывающей обратной связи действия инсулина [4, 21] , так как она является выключенной при дефекте секреции инсулина у мышей IRS2-/- [22]. Подавление экспрессии IRS2 в &beta -клетках, сопровождаясь поддерживающейся гиперинсулинемией , может, таким образом, вносить свой вклад в нарушение продукции инcулина на протяжении длительного времени. Такая идея представляется важной для изменения бытовавшего до теперешнего времени мнения, что функциональная неспособность &beta-клеток всецело является причиной возникновения (атрибутом) гипергликемии ("отравлению глюкозой") [23, 24].
Заключение
Прежде было показано, что в процессе возникновения резистенстности к инсулину, вызванную фактором некроза опухоли α (TNFα) [25], происходит фосфорилирование посредством IRS1 остатков серина в место остатков тирозина . Можно предположить, что семейство IRS может быть часто мишенью агентов, вызывающих резистентность к инсулину.
В моделях, описанных Shimomura et al. [3], продукция печенью глюкозы и жирных кислот возрастает одновременно (Рис. 1). По всей вероятности устанавливается подпитывающая положительная обратная связь, или порочный круг, приводящий в дальнейшем к инсулиновой резистентности, так как циркуляция неэфиризированных жирных кислот вероятно приводит к уменьшению ответа на инсулин клеток ткани печени и других тканей [27]. Более того, увеличение продукции печенью глюкозы будет увеличивать выделение инсулина, вызывая подавление в печени функции IRS2 и опосредованное инсулином подавление глюконеогенеза.
Как подчеркивает Shimomura et al. [3], эти результаты подтверждают предположение McGarry [28], что диабет не должен рассматриваться только как болезнь гомеостаза глюкозы, но и по крайней мере в равной степени и как дефект липидного метаболизма. Теперь главным вопросом становится определение сходства механизмов развития резистентности печени к инсулину, описаной здесь для грызунов, механизмам развития резистентности к инсулину у человека.