и их рецепторы участвуют в управлении направлением роста и фасцикуляции (образовании пучков) аксонов, образовании границ (boundary) в мозге и миграции клеток нервного гребня. Критичным для направления движения аксонов и образования эндолимфы является цитоплазматический домен
(Eph рецептор B2 тирозин киназы).
-null мыши имеют аномалии в early midline navigation (в раннем пересечении срединной линии) контралатеральных (относящихся к внутреннему уху) эфферентных ростовых конусов, а также зависимые от линии мышей аномалии поведения (гиперактивность, круговые движения) и дисфункции вестибулярного аппарата, связанные с ультраструктурными аномалиями в продуцирующих эндолимфу вестибулярных dark клетках, которые в норме экспрессируют
. У взрослых
-null мутантов обнаружен коллапс протоков полукруглых каналов, очевидно в результате уменьшения образования эндолимфатической жидкости. В соответствие с клеточно-автономной ролью в регуляции (объема) жидкости PDZ домен-содеожащие белки, связывающие С-терминали
, распознают С-терминали определенных обменных анионов и аквапоринов (например, AQP1). Эти наблюдения позволили выявить механизм, посредством которого Ephs и их рецепторы связаны с ионным гомеостазом и образованием эндолимфы во внутреннем ухе.
Меланоциты улитки являются производными интермедиальных клеток stria vascularis нервного гребня. Они необходимы для нормального развития улитки, о чем свидетельствуют исследования мутантных мышей с врожденными аномалиями меланоцитов. Более того, Na+ K+-ATPase и калиевые каналы интермедиальных клеток необходимы для образования эндокохлеарного потенциала и для подготовки ионного окружения в stria. В соответствии с этим дефицит меланоцитов, обусловленный разрушением некоторых генов, ведет к нарушениям слуха как у мышей, так и у человека. Null-мутации в гене мышей, кодирующем эндотелиновый (endothelin) рецептор типа В (
Ednrb) или его лиганд эндотелин-3 (
End3), играют ключевую роль в процессе развития двух клеточных линий, являющихся производными нервного гребня – нейронов миэнтерического ганглия и эпидермальных меланоцитов.
Ednrb и
End3 нокауты характеризуются аганглиозным мегаколоном, ассоциированным с бело-пятнистой окраской шерсти, что напоминает фенотипы естественных мышиных мутаций
piebald-lethal и
lethal-spotting соответственно. Фактически кроссбридинг подтвердил, что
Ednrb является аллелем локуса
piebald, а
End3 аллелен локусу
lethal-spotting. Ранее было показано, что
piebald-lethal мутанты характеризуются аномалиями внутреннего уха, возможно, являющимися результатом дефектов в области акустического ганглия (производного нервного гребня) или следствием отсутствия меланоцитов (производных нервного гребня) во внутреннем ухе. У человека синдром
Waardenburg-Shah (WS4; OMIM:277580) относится к аудио-пигментарному синдрому, включающему синдром Waardenburg и болезнь Hirschsprung. Он характеризуется нарушениями слуха и аномалиями пигментации кожи и радужной оболочки. К настоящему времени клонировано три гена, кодирующих WS4 –
SOX10 (SRY-box содержащий ген10),
EDN3 и
EDNRB.
Считают, что ионы К+ подвергаются рециклингу из эндолимфы через сенсорный эпителий и обратно в stria vascularis посредством передвижения от клетки к клетке через нексус (щелевидное соединение – gap junction). Предполагают, что ионы К+ выходят из OHCs через KCNQ4 (potassium voltage-gate channel, подсемейство Q, член 4) – новый К+ канал, экспрессирующийся в OHCs и мутированный при форме несиндромальной доминантной глухоты (DFNA2; OMOM:600101) у человека. После выхода из OHCs K+ может быть удален, частично путем поглощения в опорные клетки Deiters. Согласно модели рециклинга К+, К+ затем диффундирует через систему щелевидного соединения (gap junction), связывающую клетки Deiters и фиброциты, обратно в stria vascularis. Получены доказательства, что
Slc12a7 (solute carrier family 12, член 7, другое название
Kcc4) способствует сифонированию К+ ионам после того как они вышли из OHCs в клетки Deiters, где К+ проникает в путь щелевидного соединения. К Р8, до того как у мышей обнаруживается слух, ген
Slc12a7 экспрессируется в stria vascularis и почти во всех клетках кортиева органа. На P14, когда животное начинает слышать, экспрессия
Slc12a7 ограничивается клетками Deiters, которые поддерживают OHCs на их базальной опоре (basal pole) и в фалангеальных клетках (phalangeal cells), обертывающих IHCs. Во время третьей постнатальной недели развития
Slc12a7-null мыши характеризуются прогрессирующей потерей слуха и значительной дегенерацией в спиральном ганглии в присутствии неповрежденной рейснеровой мембраны. Ограниченная выживаемость OHCs в апикальном завороте улитки, возможно, обусловливает остаточный слух, выявленный у взрослых нокаутов. Следует заметить, что глухота, ассоциированная с почечноканальциевым ацидозом, указывает на роль
Slc12a7 в экструзии (вытеснении) Cl- через базолатеральную мембрану кислото-секретирующих α-интеркалированных клеток в нефрон. Данные о том, что хлоридные анионы и бикарбонат работают как инородный (несвойственный) датчик напряжения OHC моторного белка prestin, свидетельствуют о том, что изменения в [Cl-] в кортиевом органе, могут в дальнейшем привести к нарушению функций OHC у
Slc12a7-null мышей.
У человека мутации в
GJB2 гене, кодирующем мембранный канальный белок щелевидного соединения β2 (его также называют connexin 26) отвечают примерно за 50% аутосомно-рецесствной несиндромальной глухоты (DFNA3, DFNB1). GJB2 участвует в рециклинге ионов К+ обратно в эндолимфу кохлеарного протока после стимулирования сенсорных волосковых клеток.
Gjb2-null эмбрионы гибнут на Е11 из-за нарушений трансплацентарного поглощения глюкозы. Таким образом, такие таргетированные нокауты могут служить моделью для DFNB1 и/или GJB2 патологии человека. Мутация в гене GJB3 человека (gap junction membrane channel protein β3; также называется connexin 31) также вызывает глухоту. У мышей
Gjb3 инактивация ведет к эмбриональной гибели и транзиторному плацентарному нарушению морфогенеза. Однако никаких морфологических или функциональных дефектов внутреннего уха у выживших нокаутов выявлено не было.
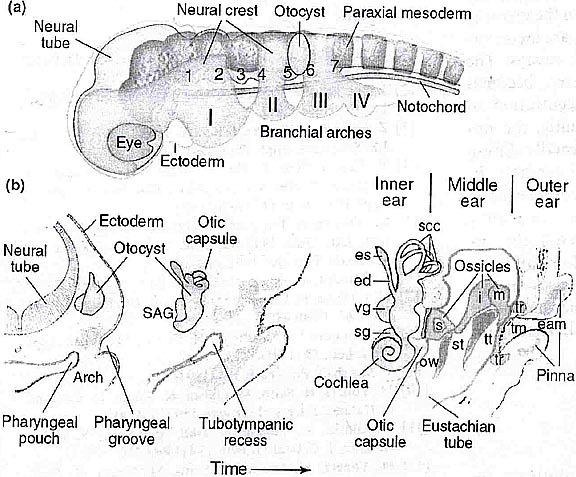
Нахождение пути аксонами и иннервация сенсорных нейронов внутреннего уха
Недавно получены доказательства того, что для выживаемости нейронов и дифференцировки сенсорных нейронов внутреннего уха необходим каскад транскрипционных факторов. Для иллюстрации этого можно привести пример с
Neurod3 (neurogenetic differentiation 3, также называется neurogenin 1), который специфически экспрессируется в VIII краниальном ганглии/нейрогенной области на стадии отоциста. Делеция
Neurod3 ведет к уменьшению сенсорного эпителия, лишению его иннервации и снижению числа волосковых клеток (которые к тому же имеют нарушения морфологии), что свидетельствует о необходимости нормальной иннервации для дифференцировки волосковых клеток, по крайней мере, до рождения. VIII ганглий у них не формируется, а макула, utricle и saccula уменьшены в размерах. Modiolus (костная канал, который образует центральную ось улитки) утрачен, а saccule образует лишь небольшое расширение на utricle, ведущее к ductus reunions, соединенному с улиткой. У
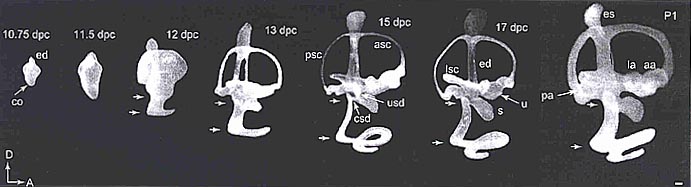
Neurod3-null мутантов имеется только 1,25 завитков вместо 1,75 по сравнению с контролем (РИС.2). Важно то, что у этих нокаутов сохранены только дистальные, эпибранхиальные, происходящие из плакоды висцеральные афференты в лицевом и stato-acoustic нервах, проецирующихся на одиночный путь (solitary tract).
Neurod1 (neurogenetic differentiation 1), нижестоящая мишень
Neurod3, также важен для выживаемости сенсорных нейронов внутреннего уха на ранних стадиях дифференцировки. Кроме двигательных дисфункций, являющихся результатом дефектов мозжечка,
Neurod1 нокауты характеризуются тяжелой атаксией и глухотой. В отличие от
Neurod3 нокаутов, у которых полностью отсутствуют проекции афферентных и эфферентных волокон,
Neurod1-нокауты еще сохраняют некоторое количество сенсорных нейронов. Паттерн оставшихся сенсорных нейронов и афферентной иннервации свидетельствуют о раннем начале такой утраты во время развития, которая стабилизируется к Е14.5. Миграция оставшихся вестибулярных сенсорных нейронов имеет неправильное направление и проекции волокон во всем вестибулярном эпителии беспорядочны. Отсутствие
Neurod1-null сенсорных нейронов, которые экспрессируют
Ntrk2 (neurotrophic tyrosine kinase, receptor, type 2, также называется
TrkB) и
Ntrk3 (neurotrophic tyrosine kinase, receptor, type 3, также называется
TrkС), указывает на участие Trk сигнализирования в выживаемости юных дифференцирующихся нейронов. Фенотип
Neurod1 нокаутов сходен с фенотипом компаундных “Trk” мутаций. Утрата иннервации в базальном завитке улитки и отсутствие спирализации афферентных и эфферентных волокон вдоль IHCs в направлении к базальному концу улитки напоминает дефекты у
Ntf3 (нейротрофин 3)-null и
Ntrk3-null мутантов. Однако паттерн утраты нейронов у
Neurod1 нокаутов больше похож на паттерн комбинированной
Ntrk2-гетерозиготной и
Ntrk3-null мутации. Такие компаундные мутации имеют высокий уровень межиндивидуальной вариабельности в плотности оставшихся афферентных и эфферентных волокон.
Функциональная делеция
Bdnf (мозговой нейротрофический фактор) или его рецептора
Ntrk2 ведет к катастрофическому снижению числа вестибулярных нейронов и полной утрате иннервации полукруглых каналов. Кроме того, у мышей с отсутствием и
Bdnf, и
Ntf3 или
Ntrk2 и
Ntrk3 утрачена вся иннервация внутреннего уха. Т.е. эти нейротрофины и их рецепторы необходимы для нормальной афферентной иннервации внутреннего уха.
Pou4f1 (POU домен, класс 4, транскрипционный фактор 1; также называется
Brn3.0 и
Brn3a) экспрессируется в facial-stato-acoustic ганглии до дифференцировки сенсорных нейронов и иннервации отоциста. Таргетирование
Pou4f1 ведет к уменьшению размеров нейронов и к снижению генной экспрессии
Ntrk3, Pva (парвальбумин) и
Pou4f2 в спиральном ганглии, указывая на то, что эти гены являются нижестоящими мишенями для
Pou4f1. Афферентная иннервация в конечном итоге устанавливается к Е13.5, несмотря на наблюдаемую задержку в образовании проекций аксонов на улитку и задний вертикальный канал. Избирательная утрата
Ntrk3 нейронов в спиральном ганглии улитки
Pou4f1-null мышей ведет к аномалиям иннервации, сходными с таковыми у
Ntrk3-null мышей. Эфферентные аксоны, использующие афферентные волокна в качестве скаффолдов («подпорок», «строительных лесов») во время нахождения своего пути, направляются к внутреннему уху по неверному маршруту. В целом, эти находки говорят о неизвестной ранее роли
Pou4f1 в контроле выживаемости, дифференцировки, миграции и нахождении аксонами сенсорных нейронов внутреннего уха пути посредством регуляции нижестоящих в сигнальном пути генов.
Постнатально
Igf1 (инсулин-подобный ростовой фактор-1) активно экспрессируется в улитке и в субпопуляции нейронов кохлеарного ганглия, а также в stria vascularis, spiral limbus и опорных клетках кортиева органа. Анализ молодых
Igf1-null мышей показал, что у них укорочена улитка и кохлеарный ганглий, незрелая покровная мембрана, и большая потеря слуховых нейронов. Утрата IGF-1 в значительной степени нарушает выживаемость клеток, их дифференцировку и созревание клеток кохлеарного ганглия, а также иннервацию сенсорных клеток кортиева органа.
Chrna9 (холинергический рецептор, никотиновый α пептид 9; также называется
Acra9) является единственной никотиновой рецепторной субъединицей, экспрессируемой кохлеарными OHCs, которые контактируют через эфферентные волокна (первоначально холинергические), происходящие из ЦНС.
Chrna9 дефицитные мыши внешне нормальны, но при выполнении рефлексов Preyer имеют отклонения равновесия и аномалии движения. Однако большинство мутантных OHCs иннервируется единственной крупной терминалью, а не множеством мелких, свидетельствуя о роли
Chrna9 в нормальных синаптических связях между эфферентными волокнами и волосковыми клетками.
Chrna9-null мыши функционально «де-эфферентированы», у них не подавлен кохлеарный ответ во время активации эфферентных волокон.
Присутствие
tenascin-C во время прокладывания маршрутов нервными волокнами в spiral lamina и в кортиев орган подтверждает, что этот гликопротеин может участвовать в афферентном синаптогенезе и служить субстратом роста нейронов в улитке, особенно для волокон, растущих в направлении OHCs области. Однако tenascin-нокауты не имеют аномалий слуха и дефектов анатомии улитки, хотя у них были отмечены некоторые отклонения в поведении. Исследования
in vitro и анализ еще одной мутантной линии нокаутов –
Tnc выявили роль tenascin-C в развитии и регенерации улитки.
Глухоту и нейродвигательные нарушения наблюдали у нокаутов, являющихся моделью метахроматической лейкодистрофии, лизосомального сфинголипидного расстройства, вызванного дефицитом
As2 (arylsulfatase) и характеризующегося прогрессирующей демиелинизацией и множественными неврологическими симптомами (OMIM:250100). Внутреннее ухо
As2 мышей почти полностью демиелинизировано и имеет дегенеративные изменения слухового ганглия, что сопровождается утратой вызванных слуховых потенциалов ствола мозга в возрасте 12 мес. Молекулярные основы специфической чувствительности мышиного слухового ганглия к демиелинизации и нейродегенерации пока неясны, т.к. эти нокауты проявляют неожиданно умеренный фенотип в центральной и периферической нервных системах.
Wnt сигнализирование участвует в контроле пролиферации и в образовании синапсов во время неврального развития и предположительно это опосредуется Frizzled семейством поверхностно-клеточными рецепторами. Экспрессия
Fzd4 (frizzled гомолог 4 дрозофилы) в слуховых и вестибулярных волосковых клетках и позднее начало потери слуха у
Fzd4 нокаутов в отсутствие гибели слуховых или вестибулярных волосковых клеток, свидетельствует, что
Fzd4 важен для поддержания функций волосковых клеток, но не играет решающей роли на ранних стадиях развития и функционирования внутреннего уха.
Наконец, обработка Slc1a3-null мышей с отсутствием solute carrier family 1, члена 3 акустически травмирующими стимулами ведет к увеличению накопления глутамата в перилимфе и ухудшению слуха из-за афферентного дендритного разрастания (возвышения) ниже IHCs. Эти исследования подтверждают, что Slc1a3, Na+-зависимый глутаматный рецептор, который специфически экспрессируется в области IHCs, и фиброциты в limbus и спиральной связке (spiral ligament), действуют как нейропротекторы против глутаматной excitotoxicity во время звукового сверхраздражения. Кроме того, это ясно свидетельствует о том, что глутамат является нейротрансмиттером синапсов между IHCs и слуховым нервом.