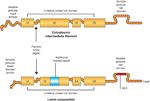
Ламина была описана как 'tensegrity element', который противостоит силам деформации и защищает хроматин от физических повреждений
. Эта гипотеза подтверждается рядом исследований. Напр., бесклеточные экстракты ооцитов
, после физического или функционального истощения ламинов, ведут к сборке очень небольших ломких ядер
. Сходным образом, RNA interference knock-down C-ламина у
. В сперматоцитах мышей ядра скорее имеют крючко-образную форму, чем яйцевидную или сферическую и эти клетки экспрессируют специфичный для зародышевой линии ламин, lamin B
. Когда lamin B
экспрессируются эктопически в соматических клетках, то их ядра адаптируют крюко-образную морфологию
. Наконец, экспрессия доминантного мутантного ламина B
, лишенного 4/5 палочковидного домена, вызывает массивную деформацию ядерной оболочки соматических клеток
. Т.о., ламины предопределяют размер, форму и прочность ядерной оболочки и , следовательно, обладают ключевыми признаками элемента tensegrity
. Однако, свойства этого tensegrity элемента, по-видимому. предопределены не только свойствами индивидуальных ламинов, но и также тем способом. которым они взаимодействуют с компонентами цитоскелета.
Недавно описано новое семейство spectrin-repeat белков, некоторые члены которого являются интегральными белками INM, а др. м. специфически локализоваться на наружной стороне ядерной мембраны. По-разному называемые nesprins
19, NUANCE
20 или synes
21, эти белки характеризуются гигантскими размерами некоторых альтернативно сплайсированных вариантов (они м. иметь относительные мол. массы, превосходящие 800,000). Они обладают множественными образованными в кластеры spectrin повторами по всему стрежню белка, N-терминальными calponin homology доменами и консервативным С-терминальным один раз пронизывающим мембрану доменом. NUANCE соединяется с актином и его распределение зависит от актинового цитоскелета
20.
In vitro, nesprin 1α - относительно ocus небольшая nesprin изоформа - соединяется с lamins A и C и с emerin, типа 2 интегральным мембранным белком INM
22. Его локализация на INM зависит от экспрессии lamins A и C
23. Nesprins также ассоциирует сам с собой
19. Кроме того, emerin непосредственно соединяется с ламинами A/C
in vitro и локализуется на INM с помощью комплекса из этих двух белков
24,25; более того, он является также актин-связывающим белком
26. Т.о., lamins A и C м. собирать комплекс белков на ядерной оболочке, которые обладают характеристиками белков, связывающихся с цитоскелетом
27 и потенциально связывают актиновый цитоскелет с ламиной. Одна из моделей предполагает, что интегральные белки INM, которые соединяются с ламинами A and C, взаимодействуют или непосредственно или косвенно посредством белков в околоядерном пространстве с крупными изоформами nesprin на наружной стороне ядерной мембраны, которые в свою очередь соединяются с элементами цитоскелета. Это м.б. тем способом, с помощью которого, когда клетки экспрессируют A-type ламины в месте своей дифференцировки, они приобретают контакты между ламиной и цитоскелетом.
Учитывая эти соединения, свойства растяжимости ламинов д. передаваться через цитоскелет плазматической мембране (Рис. 2), генерируя способность механотрансдукции сигналов в клетке, которая связана с внеклеточным матриксом, внутрь ядра
28. Имеются убедительные доказательства, подтверждающие эту гипотезу. Фибробласты от
Lmna-/- мышей были предметом механических напряжений при этом измеряли ядерную деформацию и индуцированную напряжением передачу сигналов
29. Ядра фибробластов от
Lmna-/- мышей имели заметно более высокую деформацию, чем фибробласты от
Lmna+/+ в результате это приводило к понижению жизнеспособности после механических воздействий. Кроме того, передача сигналов NF-κB ослаблена в ответ на механическую или цитокинами стимуляцию
29. В комплементарном исследовании, заручившись биомеханическим устройством, исследовали, как фибробласты от одних и тех же
Lmna-/- мышей противостоят силам компрессии
30. Устройство было способно одновременно измерять силы декомпрессии, генерируемые отдельными клетками, подвергнутых сжатию, а также наблюдать как деформируются клетки и их ядра. Ядра фибробластов
Lmna-/- мышей заметно менее способны противостоять компрессии по сравнению с ядрами фибробластов
Lmna+/+ мышей. Кроме того, ядра от
Lmna-/- мышей обнаруживают изотропную деформацию, когда сдавлены, тогда как
Lmna+/+ мыши обнаруживают анизотропную деформацию. Это указывает на то, что ядра клеток, лишенные A-type lamins более не реагируют на полярность клетки и что это отсутствие реакции происходит из-за того, что все элементы цитоскелета дизорганизованы вокруг ядерной оболочки
30. Т.о., в мезенхимных клетках, по крайней мере, mechano-индуцированная передача сигналов распространяется с помощью связи ламины с цитоскелетом.
Lamins as regulators of transcription
Ряд сообщений связывает ламины с транскрипцией и пост-транскрипционным процессингом. В ооцитах
Xenopus laevis специфический для зародышевой линии ламин Liii ассоциирует с RNA polymerase II (RNA pol II) и если он он неспособен формировать филаменты благодаря доминантно-негативной мутации lamin, то активность RNA pol II ингибируется
31. В соматических клетках lamin B
1 соединяется с POU доменом репрессорного белка Oct-1, который регулирует экспрессию гена collagenase
32. Кроме того, LAP2β, интегральный белок INM, вместе с B-type lamins, формирует функциональные комплексы с с транскрипционными факторами germ-cell-less (GCL) и DP, чтобы репрессировать E2F
33. Т.о., B-type lamins, по-видимому, оказывают негативное и позитивное влияние на транскрипцию.
A-type lamins также играют роль в метаболизме РНК. A-type lamins распределены по всей внутренности ядра и ассоциируют не только с ламиной, но и также с рядом ядерных телец. Это указывает на то, что они участвуют в транскрипции и процессинге РНК
34,35,36. Кроме того, A-type lamins ассоциируют со специфическими транскрипционными факторами и, по крайней мере, одна из этих ассоциаций является функциональной
37. A-type lamins, как известно, ассоциируют с retinoblastoma protein (Rb)
37, sterol response element binding protein 1 (SREPB1)
38 и MOK2 (ref. 39), тогда как emerin соединяется с GCL
40. C-терминальная область lamins A и C взаимодействует также с ДНК
41.
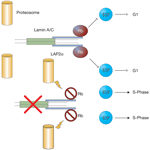
Функция взаимодействий lamin-A- или lamin-C-Rb сегодня становится ясной. В фибробластах lamin A/C и белок нуклеоскелета LAP2α формируют комплекс с Rb, который закрепляет нефосфорилированные формы белка внутри ядра
42. Закрепление Rb в ядре необходимо для его функции, а мутантные формы Rb, которые не м.б. закреплены, способствуют раку
43. Важно, что эти мутанты не взаимодействуют с lamin-A- или lamin-C-LAP2α
42. Получены прямые доказательства роли lamins A и C в функции Rb. В клетках от
Lmna-/- мышей, Rb истощен и направляется на деструкцию с помощью протеосом
10. Более того, фибробласты обладают характеристиками роста и размерами, типичными для фибробластов от
Rb-/- мышей. Т.о., причина взаимодействия lamin-A- или lamin-C-Rb, по-видимому, заключается в предупреждении отправки Rb на деструкцию с помощью протеосом, тем самым регулируется рост и деления клеток (Рис. 3).
The laminopathies: Human diseases caused by mutations in LMNA raise new questions
В начале 1990s был идентифицирован ген, ответственный за X-linked Emery-Dreifuss muscular dystrophy (X-EDMD) с помощью позиционного клонирования. Идентифицирован и новый ген, кодирующий интегральный мембранный белок, названный 'emerin'
44. Белок назван в честь Alan Emery, который вместе с Fritz Dreifuss впервые описал клиническое нарушение с контрактурами локтевых, ахиллесовых сухожилий и задней части шеи, медленно прогрессирующей мышечной слабостью и кардиомиопатией с блоком атривентрикулярного проведения
45. Позднее было установлено, что emerin локализуется на ядерной оболочке
46,47 и взаимодействует с A-type lamins
25,48. Это было первым заболеванием у людей, обнаружившим связь с INM белком. Заболевание, идентичное X-EDMD, наследуемое аутосомно доминантно (AD-EDMD). Используя позиционное клонирование др. группа показала, что AD-EDMD обусловливается мутациями в
LMNA49. Затем мутации в
LMNA были найдены и как причина родственного аутосомно-доминантного заболевания поперечно исчерченных мышц с превалирующей кардиомиопатией и с незначительными или без вовлечением скелетных мышц
50 или кардиомиопатии с вовлечением скелетных мышц поясов конечностей
51. Ещё раньше была установлена структурная организация lamin-A/C-кодирующей части
LMNA52. Большинство мутаций в
LMNA, вызывающих болезни поперечно-полосатых мышц, как было установлено, ведут к небольшим делециям или аминокислотным заменам по всему lamins A и C, с редкими мутациями, вызывающими гаплонедостаточность
49,53.
Dunnigan-type familial partial lipodystrophy (FPLD) или lipoatrophic diabetes, является аутосомно-доминантно наследуемым заболеванием, характеризующимся потерей жира из конечностей, избытком жира на лице, шее и туловище и усточивостью к инсулину
54. По многим аспектам FPLD напоминает метаболический синдром, который возникает у старых индивидов и характеризуется ожирением живота, повышенными уровнями липидов в сыворотке, высоким кровяным давлением, устойчивостью к инсулину, провоспалительным состоянием и протромботическим состоянием
55. Ген, ответственный за FPLD, сцеплен с хромосомой 1q21-1q22 (ref. 56), с областью, в которой ранее был картирован
LMNA57. Затем
58-60 было показано, что мутации в
LMNA вызывают FPLD. Примерно 90% мутаций являются missense мутациями, локализованными в экзоне 8. Аминокислотные замены меняют заряд на поверхности, экспозированной к solvent, в иммуноглобулиновой складке хвоста ламинов A и C
61,62. Нпротив, аминокислотные замены в той же самой области молекулы, которая вызывает заболевания поперечно-полосатых мышц, ведут к общему распаду структуры трёхмерной складки. Следовательно, мутации, которые вызывают FPLD вызывают лёгкие изменения молекулярной структуры lamin A/C, тогда как мутации. вызывающие болезни поперечно-полосатых мышц, вызывают более драматические структурные альтерации. Вызывающие FPLD мутации A-type lamin м. функционировать в доминантно interfering способом, т.к.
Lmna-/- мыши не дают частичную lipodystrophy
63.
Недавно было показано, что мутации в A-type lamins не только вызывают нарушения поперечно-полосатых мышц и ожирение, но и осложненный и запутанный набор заболеваний. Некоторые гомозиготные мутации
LMNA вызывают аутосомно-рецессивные нейропатии периферических аксонов у людей, а
Lmna-/- мыши обладают патологически сходными аномалиями периферических нервов
64. Гомозиготные мутации в
LMNA вызывают также mandibuloacral dysplasia (MAD), редкое нарушение, характеризующееся задержкой постнатального роста черепа и лицевыми аномалиями, скелетными уродствами, крапчатой пигментацией кожи, частичной lipodystrophy и признаками преждевременного старения
65. MAD вызывается также мутациями в ZMPSTE24 protease, участвующей в процессинге pre-lamin A в lamin A, a
Zmpste24-/- мыши имеют фенотипическое сходство с MAD людей
66-68. Недавно гетерозиготные мутации и в
LMNA и в
ZMPSTE24 обнаружили ассоциацию с рестриктивной дермопатией, известной также как 'tight-skin contracture syndrome', др. редким заболеванием, характеризующимся в основном задержкой внутриматочного роста, плотной и ригидной кожей с эрозиями, лицевыми аномалиями, дефектами минерализации костей и ранней неонатальной гибелью
69. Аутосомно-рецессивные мутации в
LMNA также вызывают синдромные нарушения с генерализованной потерей жира, резистентным к инсулину diabetes mellitus, рассеянными увеличивающимися кожными повреждениями, ожирением печени и кардиомиопатией
70.
Вообще-то наиболее драматическим фенотипом, вызываемым мутациями в
LMNA является progeria. Hutchinson-Gildford progeria syndrome (HGPS) является редким аутосомно-доминантным заболеванием с задержкой роста, аномальным развитием лица, потерей подкожного жира и преждевременным атеросклерозом, приводящим к гибели во второй декаде жизни
71. В 2003 две группы
72,73 сообщили о
de novo доминантной мутации в экзоне 11
LMNA у субъектов с HGPS. Эта мутация ведет к созданию аномального сплайс-донорского сайта РНК в экзоне 11, это к экспрессии укороченного pre-lamin A белка с делецией 50 аминокислот на С-конце. Эта мутация обнаруживается у большинства субъектов с HGPS, но и др.
LMNA мутации м.б. причиной прогерии у некоторых субъектов
72-74. 'Knock-in' мыши с мутацией сплайсинга РНК, отличающейся от той, что у людей с HGPS, также обнаруживают признаки прогерии
75. Мутации в
LMNA описаны также у субъектов с атипическим преждевременным старением, не удовлетворяющим диагностическим критериям HGPS или др. специфических синдромов
76,77.
Этот широкий спектр болезней, возникающий благодаря мутациям в A-type lamins, ставит новые вопросы о их функциях. Это указывает на то, что белки выполняют разные роли в разных соматических клетках. Некоторые мутанты, подобные тем. что вызывают прогерию, затрагивают многие типы клеток. Др. мутации, такие как, вызывающие EDMD или FPLD, по-видимому, оказывают свои эффекты относительно клеточно-специфическим образом. Усилия исследователей направлены на расшифровку клеточно-специфических функций A-type lamins и определение, как мутации вызывают ряд т.наз. ламинопатий.
The 'structural' and 'gene expression' hypotheses to explain laminopathies
Предложены две рабочие гипотезы для объяснения возникновения laminopathies. Согласно 'structural' гипотезе мутации в A-type lamins или в emerin вызывают ослабление ядерной оболочки, это предрасполагает к её повреждениям. В поперечно-полосатых мышцах особенно повреждения ядер м. способствовать гибели миоцитов и замещению их жировой и фиброзной тканью
8,25,78. Гипотеза 'gene expression' предполагает, т.к. A-type lamins являются важными регуляторами генной экспрессии, то мутации в этих белках д. менять их взаимодействия с разными генами регуляторных белков и тем самым способствовать заболеваниям в разных тканях
78-80. Сегодня имеются подтверждение обеих гипотез, общим является, что и структурная слабость и изменение генной экспрессии вносят вклад в патогенез.
The case for the structural hypothesis
Исследование фибробластов от
Lmna-/- мышей показали, что отсутствие A-type lamins делает ядерную оболочку структурно уязвимой, которая становится чувствительной к стресс-индуцирующим повреждениям, приводящим к потере клеточной жизнеспособности
29,30. Однако, хотя
Lmna-/- мыши и обладают фенотипом. сходным с EDMD людей
25,81, болезни людей вызываются мутациями в
LMNA, включая и EDMD, но не возникают у нулевых генотипов. Тем не менее имеются доказательства, что мутантные A-type lamins способствуют структурной слабости. Когда lamin A или lamin C мутации экспрессируются в модельных клеточных линиях, то они часто не способны ассоциировать соотв. образом с ядерной оболочкой и формируют агрегаты в нуклеоплазме
82,83. Более того, эти мутантные ламины м. способствовать структурным аномалиям в ядерной оболочке, которые обычно обнаруживаются в фибробластах людей с laminopathies
72,73,65,84-87. Очевидно, что эта структурная слабость транслируется в физические повреждения. Ультраструктурные иследования скелетных мышц от индивидов с AD-EDMD и X-EDMD
88,89 и скелетных мышц от
Lmna-/- мышей
81 выявляют и широко распространённые разрывы ядерных оболочек и вытекание хроматина в цитоплазму. Эти повреждения коррелируют с клеточной гибелью. Согласуются с этими находками то, что белки ядерной оболочки фибробластов от субъектов с EDMD обнаруживают заметное увеличение свойств растворимости по сравнению с нормой
90. Т.о., слабость ядерной оболочки способствует клеточной гибели. Нерешенным остаётся вопрос отсутствия мутаций в
LMNA и emerin примерно у половины субъектов с клиническим диагнозом EDMD
53. Если модель tensegrity верна, то новые гены кандидаты д.б. связаны с линкерными с цитоскелетом белками, такими как nesprins или компонентами цитоскелета.
The case for the gene expression hypothesis
Чёткие доказательства потенциального участия Rb в laminopathies. Rb не только участвует в клеточной пролиферации, но кго активность также необходима для дифференцировки мезенхимных клеток, включая скелетные мышцы
91 и адипоциты
92. Ясно, что потеря экспрессии lamins A и C нарушет функцию Rb и ведет к потере контроля роста
10. Экспрессия мутаций lamin A, которые обусловливают AD-EDMD в C2C12 миобластах, ингибирует их дифференцировку и способствует апоптической гибели клеток
93. Эти находки коррелируют с недавними сообщениями. показавшими, что рост в ранних пассажах фибробластов от индивидов с HGPS характеризуется быстрой пролиферацией, но с высокой скоростью апоптоза
85. Следовательно, потеря функции Rb м.б. ответственной за этот фенотип. В самом деле, подтверждено, что экспрессия мутаций lamin A, вызывающих AD-EDMD в C2C12 миобластах, предупреждает из дифференцировку путём разрушения LAP2α-Rb комплексов, это ведет к потере экспрессии изоформ Rb
94. Одной из проблем, связанных с этой находкой, является то, что они указывают на то, что мышечная дифференцировка д.б. нарушена на ранней стадии развития
Lmna-/- мышей. Однако, в действительности мыши развиваются нормально, но дегенерация мышечных волокон происходит после рождения
25. Т.к. C2C12 миобласты происходят из сателлитных клеток, то они лучше представляют собой модель регенерации, чем развития. Поэтому мы полагаем, что присутствие мутаций A-type lamin во взрослых стволовых клетках м. нарушать их способность регенерировать некоторые ткани, такие как поперечно-полосатые мышцы, т.к. определенные программы дифференцировки нуждаются в функции Rb. Наиболее тяжелые случаи ламинопатий м. возникать из-за того, что определенные клетки чувствительны к дегенерации благодаря ломкости ядреных оболочек, и в то же самое время стволовые клетки взрослых оказываются неспособны регенерировать поврежденные ткани из-за потери функции Rb.
REBP1, который играет критическую роль в дифференцировке адипоцитов, взаимодействует с lamin A
in vitro38. Мутации, которые вызывают FPLD, но не др. ламинопатии, снижают эффективность взаимодействий lamin-A-SREBP1. Следовательно, FPLD м. способствовать неправильная функция SREBP1 из-за потери связывания с lamin A.
A-type lamins as guardians of the soma
Мы подчеркиваем две существенные функции A-type lamins и ядерной ламины. Во-первых, они играют роль в качестве структур, несущих нагрузки, в центре клетки, которые противостоят сжатию и необходимы для распространения индуцируемых стрессами сигнальных путей. Во-вторых, ламиновые комплексы играют роль в регуляции транскрипционных репрессоров и, в частности, lamin A играет роль в поддержании функции Rb. Потеря функции A-type lamin нарушает оба пути и ведет к дегенерации и возможно к отсутствию нормальной регенерации в разных тканях и в некоторых случаях к преждевременной гибели. Т.о., A-type lamins, по-видимому, существенны для выживания в старости и д. рассматриваться как защитники тела (soma).
Сайт создан в системе
uCoz & Howard J. Worman
& Howard J. Worman