Пользователи: 
ПАЛАТОГЕНЕЗ
Генетические и эпигенетические причины расщепление нёба
Molecular Regulation of Palatogenesis and Clefting: An Integrative Analysis of Genetic, Epigenetic Networks, and Environmental Interactions Hyuna Im 1,†,Yujeong Song 1,†,Jae Kyeom Kim et al.
 Int. J. Mol. Sci. 2025, 26(3), 1382; https://doi.org/10.3390/ijms26031382
|
Palatogenesis is a complex developmental process requiring temporospatially coordinated cellular and molecular events. The following review focuses on genetic, epigenetic, and environmental aspects directing palatal formation and their implication in orofacial clefting genesis. Essential for palatal shelf development and elevation (TGF-β, BMP, FGF, and WNT), the subsequent processes of fusion (SHH) and proliferation, migration, differentiation, and apoptosis of neural crest-derived cells are controlled through signaling pathways. Interruptions to these processes may result in the birth defect cleft lip and/or palate (CL/P), which happens in approximately 1 in every 700 live births worldwide. Recent progress has emphasized epigenetic regulations via the class of non-coding RNAs with microRNAs based on critically important biological processes, such as proliferation, apoptosis, and epithelial–mesenchymal transition. These environmental risks (maternal smoking, alcohol, retinoic acid, and folate deficiency) interact with genetic and epigenetic factors during palatogenesis, while teratogens like dexamethasone and TCDD inhibit palatal fusion. In orofacial cleft, genetic, epigenetic, and environmental impact on the complex epidemiology. This is an extensive review, offering current perspectives on gene-environment interactions, as well as non-coding RNAs, in palatogenesis and emphasizing open questions regarding these interactions in palatal development.

|
Развитие черепно-лицевой области, особенно нёба, - один из самых сложных процессов в эмбриогенезе позвоночных, требующий точной координации многочисленных клеточных и молекулярных событий. Вторичное формирование нёба имеет решающее значение, поскольку его нарушение приводит к расщелине губы и/или нёба (CL/P) - распространенной врожденной аномалии, встречающейся примерно у 1/700 живорожденных во всех популяциях. Развитие нёба - это сложный процесс, требующий от клеток нервного гребня координации клеточных процессов, таких как рост, движение, дифференцировка и гибель клеток. Палатогенез контролируется консервативными сигнальными путями, такими как TGF-β, BMP, FGF, WNT и SHH. Эти сигнальные пути помогают нёбной пластинке расти, подниматься и срастаться, подобно другим системам органов. Эти процессы работают как часть сети, которая контролирует гены и эпигенетику. Достижения молекулярной биологии позволили связать эпигенетические процессы, включая метилирование ДНК, модификации гистонов и не-кодирующие РНК, с ключевыми аспектами развития нёбной пластинки. Когда женщина на ранних сроках беременности подвергается воздействию таких факторов, как курение, алкоголь, ретиноевая кислота (RA) и некоторые тератогены, они могут изменить генетические и эпигенетические процессы, которые влияют на развитие нёба. Очень важно понять их сложное взаимодействие, чтобы разработать более эффективные профилактические и терапевтические стратегии для орофациальных расщелин (OFCs) .
2. Craniofacial Development: Molecular and Genetic Basis
2.1. Anatomical Development and Classification of Cleft Lip and/or Palate (CL/P)
2.1.1. Anatomical Overview of Palate Formation
Твердое нёбо (костная часть спереди) и мягкое нёбо (мускулистая часть сзади) разделяют ротовую и носовую полости во время эмбриогенеза. Во время развития нёба створки поднимаются, соприкасаются и срастаются по средней линии, образуя твердую (переднее) и мягкую (заднее) часть нёба. Этот процесс необходим как для речи, так и для глотания. Сращивание нёбных пластин начинается сзади и продвигается кпереди. Развитие лица начинается на ранних сроках беременности [1]. В процессе развития формирование лица и нёба требует пространственно-временной координации различных клеточных процессов, включая, в частности, рост, миграцию, дифференцировку и апоптоз [2]. Развитие нёба - очень сложный процесс, который регулируется транскрипционными факторами, факторами роста, сигнальными молекулами и эпигенетическими регуляторами. Нарушение тонкой настройки может привести к таким черепно-лицевым дефектам, как CL/P. На этот процесс может серьезно повлиять воздействие окружающей среды, например, курение матери, зависимость от лекарств или токсинов из окружающей среды, что приводит к развитию этих врожденных аномалий [1].
Верхняя губа, philtrum и первичное нёбо развиваются из слияния медиальных носовых и верхнечелюстных отростков (рис. 1А) [3]; однако нарушение этих процессов может привести к CL/P. Вторичное нёбо сливается с первичным нёбом в его передней части и носовой перегородкой в его переднедорсальной области, причем оба развиваются одновременно (рис. 1B-D). Это слияние формирует полное нёбо в ротовой полости и отделяет его от носовой полости (рис. 1E). Нарушения поднятия нёбных отростков, контакта или нарушение их слияния приводят к вторичной расщелине нёба CP . У человека развитие нёба начинается примерно на 6-й неделе беременности и завершается к 12-й неделе [1]. У мышей этот процесс начинается примерно на ст. E11.5 и завершается к E15.5 (рис. 1) [2,3]. Сложный процесс формирования нёба зависит от точной пространственно-временной регуляции множества факторов, таких как факторы транскрипции, факторы роста, сигнальные молекулы и эпигенетические факторы, для нормального развития. Нарушение этого процесса вследствие курения матери, злоупотребления лекарственными препаратами или воздействия факторов окружающей среды может привести к черепно-лицевым деформациям с CL/P Все эти факторы находятся в сложном равновесии для правильного формирования и слияния нёбных пластинок. Небольшое нарушение этого тонкого баланса приводит к аномалиям развития, часто к ЦП, и другим черепно-лицевым аномалиям. Понимание молекулярных механизмов, участвующих в формировании нёба, необходимо для разработки эффективных профилактических стратегий и методов лечения нарушений OFC [3].
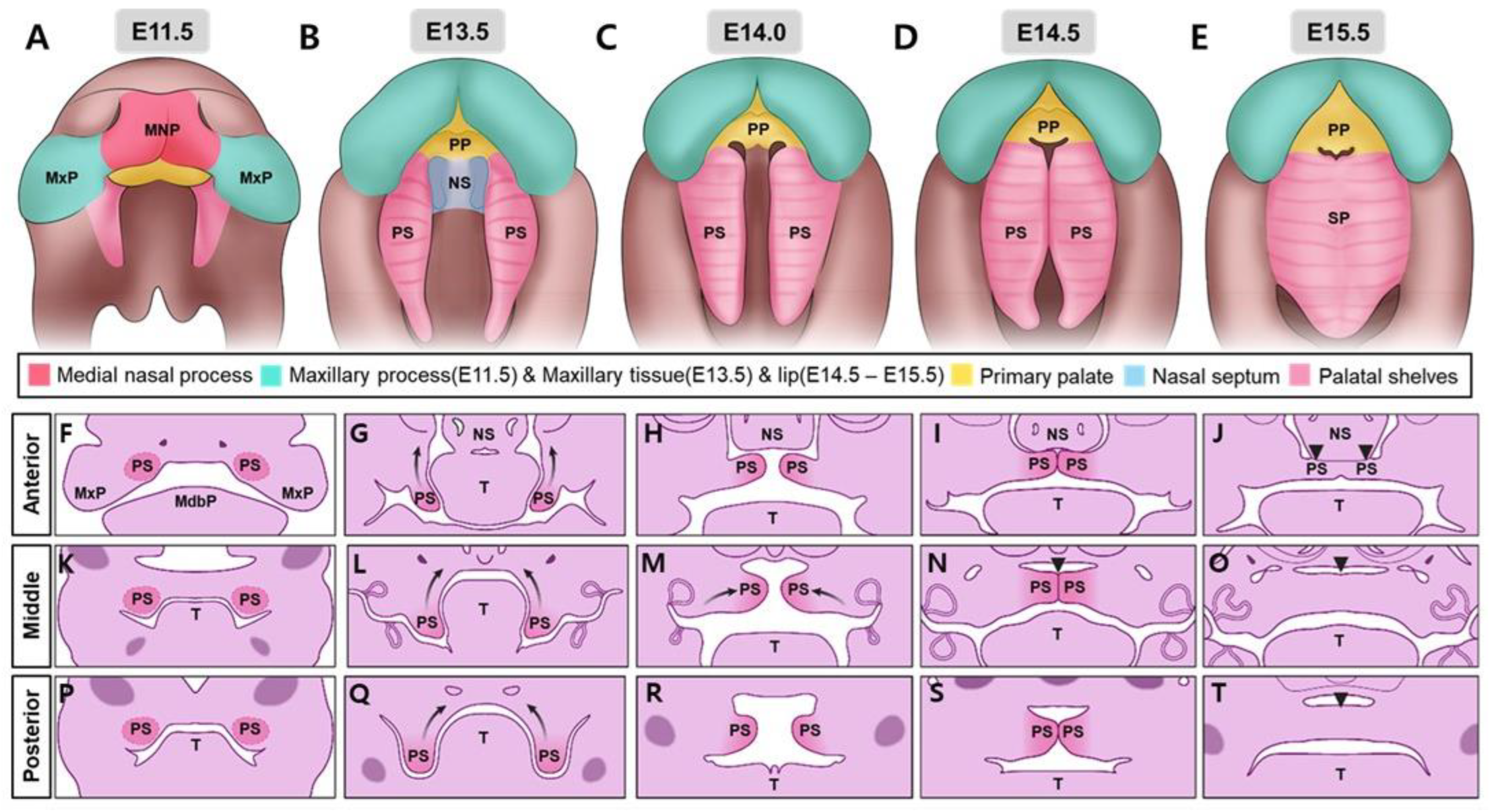 Figure 1. Developmental progression of secondary palate formation in mouse embryos from E11.5 to E15.5. (A–E) Frontal views show the developmental sequence of palatal shelf elevation and fusion. At E11.5 (A), the medial nasal process (MNP) and maxillary processes (MxP) are visible with the initial formation of the primary palate (PP). From E13.5 to E15.5 (B–E), the palatal shelves (PS) undergo vertical-to-horizontal elevation, with concurrent development of the nasal septum (NS). Progressive fusion of the PS occurs, resulting in the formation of the secondary palate (SP) by E15.5. (F–T) Frontal sections through the developing palate’s anterior, middle, and posterior regions at corresponding developmental stages. The sections demonstrate the progressive growth, elevation, and fusion of the PS around the tongue (T). The medial edge epithelium (MEE) is visible in anterior sections at early stages. The dynamic process of palatal shelf reorientation and fusion proceeds in an anterior-to-posterior sequence, with complete fusion achieved by E15.5. Color code: pink—medial nasal process (MNP); turquoise—maxillary process (MxP) (E11.5) and maxillary tissue (E13.5–E15.5) and lip; yellow—primary palate (PP); light blue—nasal septum (NS); light pink—palatal shelves (PS); black arrowheads indicate the gap between the primary and secondary palates, which will close following fusion between these tissues. Also, the black curved arrows indicate the direction of palatal growth.
2.1.2. Classification of Human Cleft Lip and/or Palate
CL/P - это распространенный врожденный дефект, который отличается расположением и степенью выраженности CP. CL возникает, когда ткани верхней губы не срастаются должным образом во время развития плода, в результате чего образуется щель или отверстие с одной (одностороннее) или с обеих сторон (двустороннее) (рис. 2) [1].
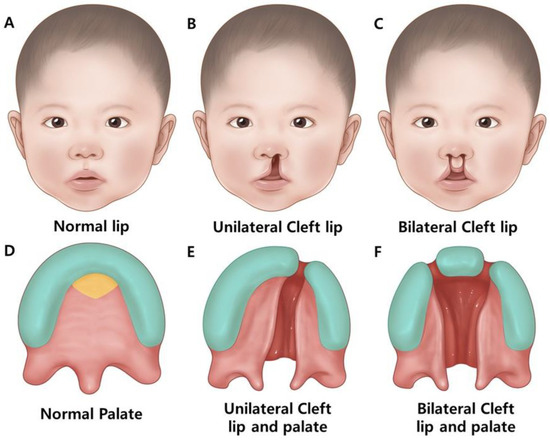 Figure 2. The anatomical spectrum of normal and cleft lip and/or palate malformations in humans. (A–C) Frontal facial views show variations in lip formation. (A) Normal lip morphology with complete fusion. (B) The unilateral cleft lip is showing incomplete fusion on one side. (C) Bilateral cleft lip presenting incomplete fusion on both sides of the upper lip. (D–F) Oral views of the palatal region depicting normal and cleft phenotypes. (D) Normal palate showing complete fusion of palatal shelves. (E) Unilateral cleft lip and palate with the incomplete fusion of the palatal shelf on one side. (F) Bilateral cleft lip and palate showing incomplete fusion of palatal shelves on both sides. Color code: turquoise—maxillary tissue; yellow—primary palate; pink—palatal and soft tissues.
Существует три подтипа односторонней расщелины губы (CL).
-
Неполная CL (меньшая щель);
--
Полная CL (полная ширина верхней губы);
-
срединная CL (редко - середина верхней губы).
Двусторонняя CL встречается реже и сложнее поддается лечению из-за тяжести деформации.
CP - это деформация крыши (roof) рта, вызванная неправильным сращением верхнечелюстных нёбных отростков на ранних стадиях беременности, в результате чего формируется либо полное (твердое или мягкое), либо неполное нёбо (рис. 2).
-
Полный CP включает в себя как твердое, так и мягкое нёбо.
-
Неполная CP охватывает и твердое, и мягкое нёбо.
-
Подслизистая CP включает в себя небольшое отверстие в мягком нёбе, при этом слизистая оболочка остается неповрежденной [1].
При подслизистой расщелине имеется дефект мягкого неба, однако вышележащий слой слизистой оболочки не поврежден. Диагностика может быть отложена до появления проблем с речью или слухом. Расщелина губы и неба (CLP) часто встречается вместе (рис. 2) [1].
Асимметричные расщелины, затрагивающие одну или обе стороны губы или нёба, могут повлиять на классификацию и лечение [4]. Хотя данные по асимметрии OFC на мышиных моделях ограничены, формирование черепно-лицевых структур может регулироваться генетическими, эпигенетическими и экологическими факторами, аналогичными тем, что существуют у человека. Несмотря на то, что было выявлено множество генетических причин и мутаций, связанных с OFC пробелы в знаниях о клеточных и молекулярных механизмах, вовлеченных в процесс, означают, что клиническая помощь и даже стратегии профилактики не претерпели значительных изменений [5]. Дальнейшие исследования с использованием мышиных моделей необходимы для разработки эффективных методов лечения.
3. The Pathogenesis of Orofacial Clefts in Humans Involves Genetic and Environmental Factors
3.1. Overview of Syndromic/Non-Syndromic Associated with Cleft Lip and/or Palate
CL/P - наиболее распространенный врожденный черепно-лицевой порок [6]. Распространенность CL составляет примерно 1 на каждые 700 живорожденных (табл. 1) [1,6]. Во всей популяции мужчин, родившихся с CL, значительно больше, чем женщин [7]. Вариации встречаемости демонстрируют значительные различия между различными расовыми и этническими группами, при этом среди афроамериканского населения заболеваемость ниже (примерно 0,5/1000) [7]. CL содержит одну или несколько расщелин, которые простираются от верхней губы до одной или обеих ноздрей. CP - это тип расщелины, которая образуется на своде рта, верхней губе или обеих губах, когда кости не срастаются должным образом в процессе эмбриогенеза. Она может затрагивать одну сторону, обе стороны или центральную область (рис. 2) [1]. Хотя CL/P не приводит к летальному исходу, пациенты с CL/P страдают от стоматологических, окклюзионных, функциональных и эстетических проблем, а также от вторичных осложнений, таких как проблемы со слухом, дыханием и питанием. Этиология CP многофакторна и включает в себя как генетические, так и экологические компоненты [1]. Мутации в различных генах, включая моногенные нарушения и хромосомные перестройки, наблюдаются у пациентов с CLP и присутствуют в многочисленных генетических синдромах (табл. 1 и табл. 2) [8].
Table 1. Comprehensive genetic information of non-syndromic orofacial clefts: Prevalence, subtypes, and associated factors.
Table 2. Genetic syndromes associated with orofacial clefts and their key features.
При наличии или отсутствии CP/CL обычно делятся на изолированные (несиндромальные) и менделевские синдромальные формы. Несиндромальная расщелина губы и/или нёба (НРГНН) (93-97 % случаев) имеет сложную этиологию, которая объясняется взаимодействием генов и факторов окружающей среды (табл. 1). Риск рецидива NSOFC оценивается как 4-10. Синдромные формы CL/P составляют около 5-7 % случаев, охватывают более 200 различных состояний и отличаются различной структурой и распространенностью врожденных пороков развития [9].
3.2. The Genetic and Epigenetic Basis of Craniofacial Abnormalities: Non-Syndromic and Syndromic Forms
3.2.1. Non-Syndromic Craniofacial Anomalies
Несиндромные орофациальные расщелины NSOFC составляют 70 % всех расщелин и являются одними из самых распространенных врожденных пороков развития человека. Их распространенность в мире составляет примерно 1 на 700 живорожденных (табл. 1) [8]. Несиндромальные расщелины, определяемые как изолированные деформации, составляют около 45 % случаев CP и 85 % случаев NSCLP (табл. 1) [9]. NSOFC ассоциируется с различными генетическими факторами, такими как однонуклеотидные полиморфизмы (SNPs), мутации отдельных генов, факторы окружающей среды и паттерны микроРНК (miRNA) (табл. 1) [9]. Путь WNT является критическим для развития черепно-лицевой области, и гены пути WNT, включая AXIN1и WNT9B, были ассоциированы с NSOFC [9,10]. Гены этого пути, в частности FGFR1 и FGF2, ассоциированы с NSOFC [9]. Другие гены, ассоциированные с NSOFC по результатам исследований сцепления, включают COL11A1, IRF6, EGF, MSX1, PTCH, TGFB1, ROR2, FOXE1, TGFB3, RARA, APOC2, BCL3 и PVRL2 [9]. Анализ связей показал, что более 20 хромосомных регионов сцеплены с NSOFC. К ним относятся хромосомы 1p, 1q21, 1q32-42.3, 6p, 2p, 4q и 17q [9].
Было проведено несколько эпигенетических широкомасштабных ассоциативных исследований (EWAS), посвященных OFC (табл. 1). В исследовании, проведенном в Великобритании, EWAS проводилось с использованием крови и тканей губы для проверки ассоциации между метилированием в каждом сайте и подтипом расщелины (расщелина только губы (CLO) n = 50; расщелина только нёба (CPO) n = 50; CLP n = 50) [11]. Недавно в работе [12] было показано, что восемь генов [12] (ABCB1, ALKBH8, CENPF, CSAD, EXPH5, PDZD8, SLC16A9 и TTC28) постоянно экспрессируются в соответствующих тканях черепно-лицевой области мыши и человека во время формирования лица, а три гена (ABCB1, TTC28 и PDZD8) демонстрируют статистически значимые ограничения по мутациям. Эти результаты подчеркивают роль редких вариантов в определении генов-кандидатов для NSOFC
3.2.2. Syndromic Craniofacial Anomalies
Хотя синдромальные OFC чаще объясняются одной врожденной причиной или нарушенным геном, чем NSOFC при определении основных механизмов возникают другие проблемы [9,16]. Этиологию, лежащую в основе фенотипа OFC, может быть сложнее определить для многих синдромальных состояний, при которых расщелины выражены слабо или редко. Часто может существовать множество генов и факторов, вызывающих заболевание, и роль конкретных генов в случаях, связанных с расщелинами, может быть описана не до конца, особенно если расщелины являются второстепенным признаком и не являются основным объектом исследований при определенных условиях [16]. Кроме того, многие синдромы обусловлены делециями, нарушающими работу нескольких генов, что еще больше усложняет ассоциацию между конкретными локусами и сращением губы и/или нёба (табл. 2) [16].
22q11.2 Microdeletion Syndrome
Синдром микроделеции 22q11.2 - это врожденное заболевание с широкой фенотипической картиной, которое возникает преимущественно в результате микроделеции хромосомы 22 в месте, известном как 22q11.2 [13]. Более чем у половины пациентов с 22q11.2DS (синдром Ди Джорджа/вело-кардио-фациальный синдром) наблюдаются черепно-лицевые пороки развития, среди которых наиболее часто встречается CP . Многие аномалии развития, наблюдаемые при этом синдроме, могут быть связаны с уменьшением числа копий генов, расположенных в удаленном регионе 22q11.2, включая возможную аберрантную экспрессию T-box TF (TBX1), роль которого в палатогенезе хорошо документирована [14]. Потеря и приобретение функции Tbx1 позволяет предположить, что дозировка Tbx1 является критической детерминантой нормального палатогенеза. Tbx1 регулирует черепно-лицевое развитие через miR-96-5p, которая подавляет экспрессию Tbx1 путем связывания с 3'-UTR его мРНК [14,15].
Van der Woude Syndrome
Синдром Ван-дер-Вауде (VWS) - наиболее распространенная форма синдромальной расщелины, составляющая около 2 % всех случаев CL/P, с распространенностью 1/34 000 живорожденных. VWS - это аутосомно-доминантное заболевание, при котором у пораженных людей наблюдается одно или несколько из следующих проявлений: CL, CP, гиподонтия или парамедианная ямка нижней губы [16]. В данном обзоре анализ семьи VWS выявил SNP, rs539075, расположенный в интроне 2 гена кадхерина (CDH2), который может быть связан с CL/P [17]. Эта группа также выявила интронный вариант NOL4 у пациентов с VWS, который ко-сегрегировал с CL/P [14].
Stickler Syndrome
Синдром Стиклера (STL) - это гетерогенное заболевание, характеризующееся аномалиями коллагена (в частности, коллагена II, IX и XI типов). Распространенность синдрома Стиклера оценивается как 1:7500-9000 [19]. STL - это заболевание, включающее врожденную близорукость, возможно, сосуществующую с катарактой, повреждение сетчатки, расщелину язычка, подслизистую расщелину нёба (SMCP), изменения в черепно-лицевой структуре, изменения в суставах и костях, гипермобильность суставов и прогрессирующую потерю слуха [19].
Молекулярная диагностика STL основана на наличии патогенных вариантов в шести генах коллагенового типа (COL2A1, COL11A1, COL11A2, COL9A1, COL9A2 и COL9A3) и двух не=коллагеновых генах (LRP2 и LOXL3) [20,21], с преобладанием аутосомно-доминантного типа наследования. Мутации в COL2A1, расположенном на хромосоме 12 (12q13.11), могут вызывать мутации SLT1 [19]. STL2 возникает из-за мутаций в гене, кодирующем α1-цепь коллагена XI в COL11A1, который расположен на коротком плече хромосомы 1 (1p21.1). Основной причиной синдрома Стиклера III типа (STL3) и неокулярного синдрома Стиклера являются делеции гена COL11A2, расположенного на хромосоме 6 (6p21.3), который кодирует α2-цепь коллагена XI типа [20].
Pierre Robin Syndrome
Синдром Пьера Робина (PRS) обозначает набор характерных черепно-лицевых фенотипов, которые обычно наблюдаются: глоссоптоз, CP, микрогнатия и обструкция верхних дыхательных путей [16]. Синдром Пьера Робена встречается у 1/8500-1/14 000 новорожденных [22]. Расщелина неба связана с делециями в 2q и 4p, а также дупликациями в 3p, 3q, 7q, 8q, 10p, 14q, 16p и 22q [22]. Доминантная модель предполагает, что гипоплазия нижней челюсти приводит к сильно ретропозиционированному языку, блокирующему подъем нёбных пластинок и дыхательные пути. В качестве альтернативы внутриутробная компрессия нижней челюсти или задержка нервно-мышечного развития могут ограничивать рост нижней челюсти и языка, что приводит к обструкции нёба и дыхательных путей [22]. Изолированный PRS был связан с мутациями в гене HMG box 9 (SOX9), связанном с SRY, или рядом с ним [23]. Также сообщалось, что мутации в гене BMPR1B вызывают PRS в двух не-родственных семьях [24]. Несколько генов, кодирующих компоненты ECM и белки, взаимодействующие с ECM, также были связаны с синдромами, включая расщелины [16].
Kabuki Syndrome
Синдром Кабуки - это генетическое заболевание, характеризующееся в первую очередь выраженными чертами лица, включая гипоплазию средней части лица, широкий кончик носа, удлиненные пальпебральные щели и большие аномальные мочки ушей. Среди других признаков - расщелина или высоко поднятое нёбо, задержка роста, когнитивные нарушения и врожденные пороки сердца [25,26]. Синдром Кабуки вызывается мутациями в гене KMT2D, который кодирует гистонную метилазу H3K4, способствующую активной транскрипции генов, или в гене KDM6A, Х-сцепленной гистоновой деметилазы H3K27. Примерно в 60-70 % случаев причиной заболевания являются мутации KMT2D [25]. Этот мутировавший ген был первым патогенным геном, признанным при синдроме Кабуки, и также известен как MLL2 [27]. В KMT2D выявлено более 50 мутаций, включая нонсенс, миссенс, сдвиг рамки считывания, небольшие делеции и варианты сайтов сплайсинга [26].
Wolf–Hirschhorn Syndrome
Синдром Вольфа-Хиршхорна (WHS) - это нарушение развития, характеризующееся умственной отсталостью, задержкой роста, пороками сердца и скелета, судорогами, а иногда и проблемами средней линии, такими как CP и асимметрия лица [16]. WHS - редкий синдром делеции смежных генов (распространенность 1:20 000-50 000 рождений, соотношение женщин и мужчин 2:1), вызванный отсутствием дистальной части короткого плеча хромосомы 4 [28]. Как правило, WHS возникает из-за делеций в хромосоме 4p16.3, которые различаются по размеру и положению. Генетические дефекты обычно представляют собой частичные делеции дистального короткого плеча хромосомы 4, но фенотип WHS также может быть обусловлен сложными хромосомными перестройками, такими как транслокации или кольцевые хромосомы [28]. Несбалансированные транслокации могут быть de novo или унаследованы от родителя со сбалансированной перестройкой. Наиболее часто наблюдаются транслокации (1) с перестройкой t(4p;8p), t(4p;7p), t(4p;11p), t(4p;20q), t(4p;21q) и t(4p;12p); (2) инвертированные дупликации, связанные с терминальными делециями в том же плече 4p; или (3) несбалансированные перицентрические инверсии [28,29].
CHARGE Syndrome
Синдром колобомы, сердца, атрезии хоан, задержки роста и развития, аномалий половых органов, аномалий ушей и потери слуха (CHARGE) - это сложное генетическое заболевание, затрагивающее множество систем организма. Синдром CHARGE - это синдром множественных врожденных пороков развития, распространенность которого при рождении оценивается как 1 на 15 000-17 000 новорожденных [16]. В большинстве случаев причиной являются мутации в ремоделировании хроматина и регуляторе экспрессии генов CHD7. CHD7, расположенный на хромосоме 8, отвечает за синдром CHARGE и был впервые обнаружен в ходе исследования, выявившего мутации в CHD7 у людей с этим заболеванием [30]. Это приводит к появлению белка, играющего роль в ремоделировании хроматина, что важно для экспрессии генов во время процессов развития. Нарушение эмбрионального развития из-за мутаций в CHD7 приводит к сложному фенотипу, затрагивающему различные органы [30].
Apert Syndrome
Синдром Аперта (AS) - один из самых распространенных синдромов краниосиностоза во всем мире. Распространенность AS составляет 1/65 000 в общей популяции [31]. У пациентов с Apert Syndrom могут развиваться такие проблемы полости рта, как тяжелая гипоплазия верхней челюсти, CL и CP [32]. CP встречается примерно у 30 % пациентов с синдромом Аперта, причем расщелины мягкого нёба встречаются чаще, чем расщелины твердого нёба [33]. AS следует доминантному генетическому паттерну, и большинство пациентов - это случаи de novo, вызванные мутациями в FGFR2 [34]. FGFR2 является рецептором для фактора роста фибробластов (FGF) и кодируется геном в локусе 10q26. FGFR2 активируется при связывании с FGF и играет роль в клеточной пролиферации, ангиогенезе и дифференцировке костей [35]. Мутация экзона IIIa FGFR2 может вызывать AS, поскольку приводит к увеличению скорости дифференцировки костной ткани мезенхимными стволовыми клетками (МСК) и развитию краниосиностоза. Распространенными типами мутаций гена являются мутации FGFR2 p.Ser252Trp (S252W) 755C > G и p.Pro253Arg (P253R) 758C > G. Мутация S252W FGFR2 обычно сопровождается тяжелыми скелетными деформациями черепно-лицевой области и более высокой частотой CP, а мутация P253R FGFR2 часто сопровождается более выраженной синдактилией рук и ног [36].
3.3. Key Genes Involved in Craniofacial Development
3.3.1. Morphological and Molecular Control of Palatal Shelf Growth and Patterning
У человека смыкание губных половин и слияние нёбных дужек происходит на 6 и 12 неделях беременности [37]. Из-за этих точных сроков животные модели, в частности мыши, очень важны для изучения черепно-лицевого развития. [2,3]. Мыши являются ключевыми модельными организмами для изучения расщелины губы и нёба, поскольку они генетически схожи с человеком и имеют схожие процессы развития лица [3].
Нёбные отростки в основном состоят из мезенхимы, полученной из нервного гребня [38]. Тонкий слой эпителия ротовой полости окаймляет их с четкой передне-задней осью (A-P). У мышей эмбриональное развитие нёба начинается примерно на ст. E11.5 (рис. 1A). Хотя клетки нервного гребня (NCC) начинают мигрировать в установленные места, нёбо, фронтоназальные выступы и нёбные половинки еще не развиты. В возрасте E12.5 не наблюдается явного формирования фронтоназальных выступов и нёбных половин (рис. 3A,B). К E13.5 нёбные половинки и вертикальные тканевые пластинки нёба начинают увеличиваться и двигаться друг к другу (Рисунок 1B и Рисунок 3C,D). В конечном итоге они сливаются по средней линии между E13.5 и E15.5 (Рисунок 1С-E и Рисунок 4) [2]. Рост и слияние этих половинок регулируется взаимодействием между эпителиальными и мезенхимными тканями вдоль передне-задней оси [39-41].
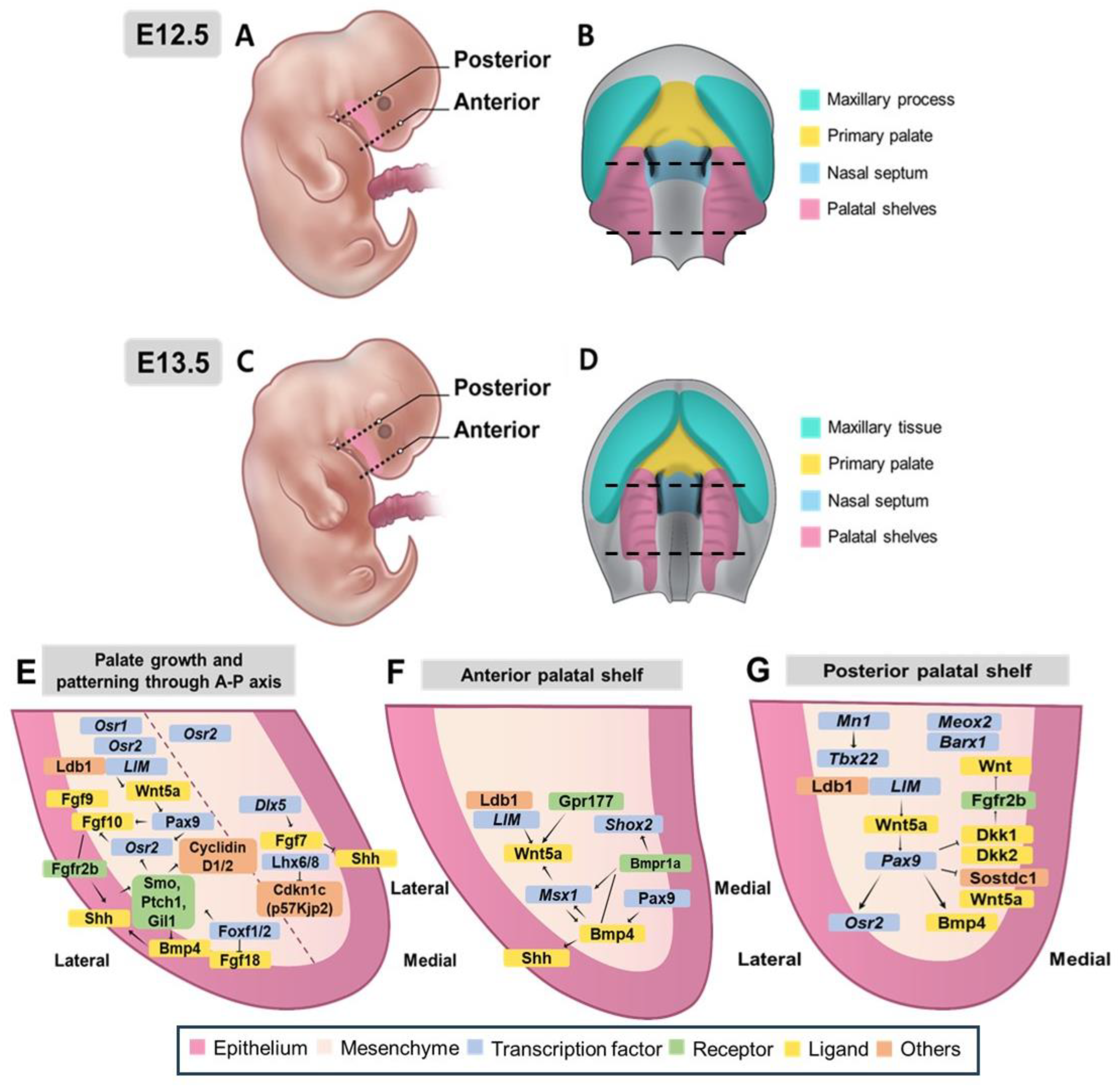 Figure 3. Complex molecular networks that coordinate palatal shelf growth, patterning, and morphogenesis along both the anterior–posterior and medial–lateral axes during palatogenesis. (A–D) Lateral and oral views of developing mouse embryos at E12.5 and E13.5 with corresponding schematic diagram The anterior and posterior orientation is indicated by dashed lines. (E–G) Schematic representations of molecular networks controlling palate development: (E) Key factors involved in palate growth and patterning along the anterior–posterior axis, showing interactions between epithelial and mesenchymal factors and their downstream targets. (F) Molecular regulation of anterior palatal shelf development. (G) Posterior palatal shelf patterning network showing interactions. Color code: pink—palatal epithelium; apricot—mesenchyme; blue—transcription factors; green—receptors; yellow—ligands; orange—other regulatory molecules.
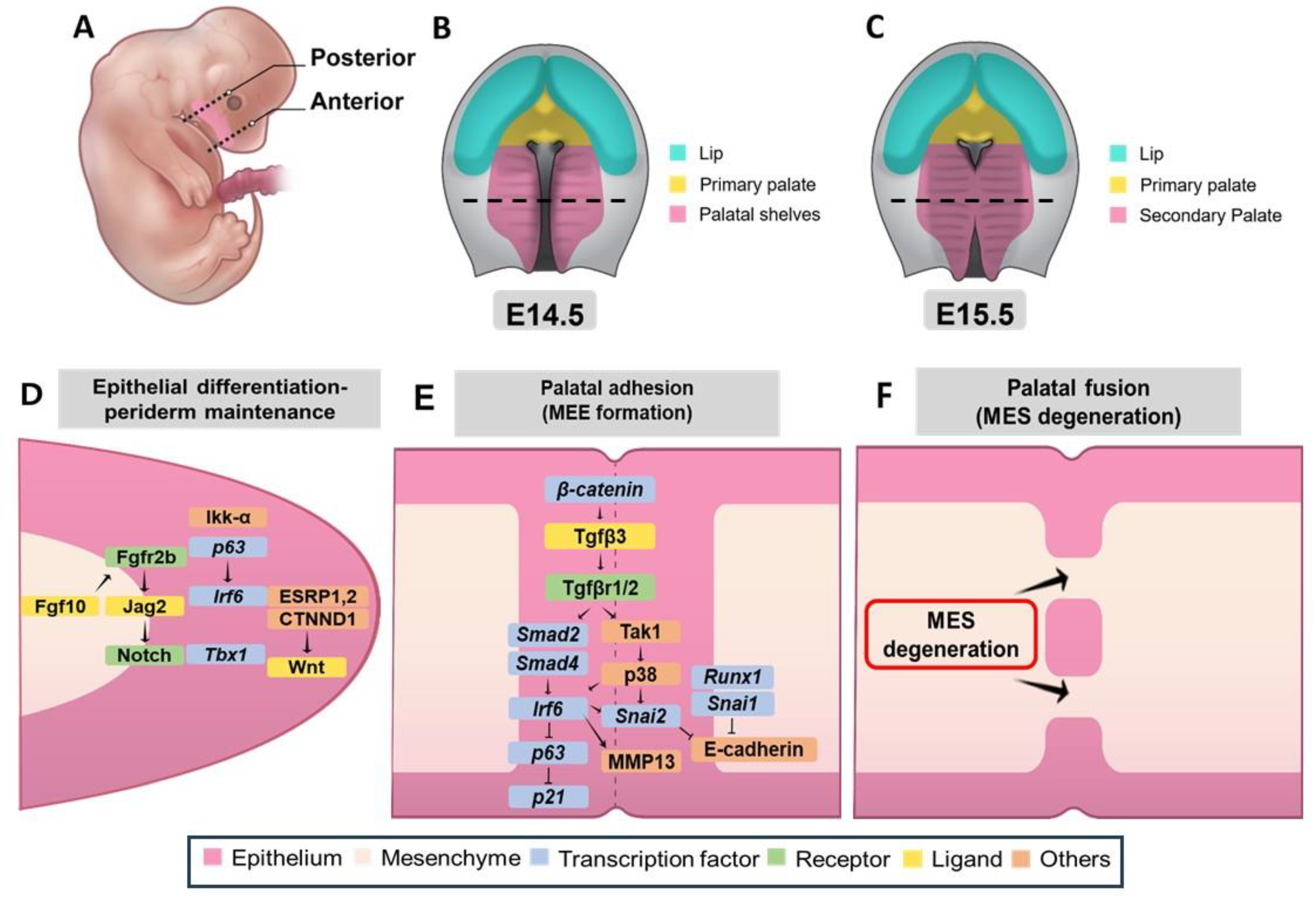 Figure 4. Development and molecular regulation of palatal shelf adhesion and fusion. (A–C) Schematic representation of mouse embryo development from E14.5 to E15.5. (A) Lateral view of E12.5 mouse embryo showing the anterior–posterior axis of palatal development. (B) Oral view at E14.5 showing elevated palatal shelves before fusion. (C) Oral view at E15.5 showing palatal fusing. (D–F) Molecular pathways controlling three key stages of palatal fusion. (D) Epithelial differentiation and periderm maintenance pathway showing genetic interactions. (E) Palatal adhesion and medial edge epithelium (MEE) formation pathway involving β-catenin, Tgf-β3, and downstream effectors. (F) Midline epithelial seam (MES) degeneration process leading to palatal fusion. Color code: pink—epithelium; apricot—mesenchyme; blue—transcription factors; green—receptors; yellow—ligands; orange—other regulatory molecules; black dotted line—remaining MES during palatal fusion. Figure 4. Development and molecular regulation of palatal shelf adhesion and fusion. (A–C) Schematic representation of mouse embryo development from E14.5 to E15.5. (A) Lateral view of E12.5 mouse embryo showing the anterior–posterior axis of palatal development. (B) Oral view at E14.5 showing elevated palatal shelves before fusion. (C) Oral view at E15.5 showing palatal fusing. (D–F) Molecular pathways controlling three key stages of palatal fusion. (D) Epithelial differentiation and periderm maintenance pathway showing genetic interactions. (E) Palatal adhesion and medial edge epithelium (MEE) formation pathway involving β-catenin, Tgf-β3, and downstream effectors. (F) Midline epithelial seam (MES) degeneration process leading to palatal fusion. Color code: pink—epithelium; apricot—mesenchyme; blue—transcription factors; green—receptors; yellow—ligands; orange—other regulatory molecules; black dotted line—remaining MES during palatal fusion.
Сигнализация SHH имеет решающее значение для роста нёбных полок, поскольку она регулирует клеточную пролиферацию и способствует развитию нёба [40]. SHH взаимодействует с другими сигнальными путями, такими как FGF и BMP, для обеспечения правильного палатогенеза. Инактивация Shh в эпителии или мезодермально-специфическая инактивация smoothened (Smo) может нарушить пролиферацию и рост нёбных клеток (рис. 3E) [42]. Кроме того, у мышей с совместными мутациями в Hhat и Ptch1 наблюдается нарушение градиента Shh во время развития фронтоназального выступа, что приводит к гипоплазии центрального и латерального выступов, в конечном итоге приводя к образованию CL и остаточных эпителиальных стыков по средней линии [43]. Первичные реснички - это небольшие волосовидные выступы, отходящие от поверхности различных тканей [44]. Необходимы для передачи сигнала Shh. Снижение экспрессии forkhead box F1 (Foxf1) в нёбной мезенхиме указывает на то, что первичные реснички являются нижестоящими эффекторами Shh-сигнализации (рис. 3E) [45].
Он необходим для роста нёбных половинок, а его отсутствие приводит к CP из-за нарушения пролиферации [46]. Хотя Fgf10 экспрессируется в мезенхиме, его рецептор, Fgfr2b, имеет решающее значение в эпителии, а эпителий-специфическая делеция Fgfr2 приводит к CP (рис. 3E) [47]. Сигнализация Shh, которая зависит от Fgf10, снижена у эмбрионов Fgf10-/- и Fgfr2b-/-, что указывает на положительную обратную связь между сигнализацией Shh и FGF, которая регулирует пролиферацию нёба [46,48]. Эти два сигнальных пути и транскрипционные факторы работают вместе, активируя мезенхимную сигнализацию, чтобы обеспечить правильный палатогенез и формирование ротовой и носовой полостей. Fgf10 поддерживает экспрессию Shh в нёбном эпителии, тогда как Fgf7 подавляет экспрессию Shh, которая регулируется Dlx5 (рис. 3E) [49]. Недавние исследования продемонстрировали сложную регуляторную сеть с участием Shh, Foxf1/2 и Fgf18 в развитии нёбных пластинок (рис. 3E). Абляция Foxf1 и Foxf2 у эмбрионов мыши препятствует росту нёбных дужек, что влияет на экспрессию Fgf18 и Shh [41]. Эта координация между транскрипционными факторами и лигандами FGF, которая контролируется сигнализацией Shh, регулирует рост и формирование нёбных пластинок (рис. 3E).
Fgf9, критический лиганд FGF в черепно-лицевом развитии, экспрессируется в нёбном эпителии и мезенхиме во время нёбного развития у мышей (рис. 3E) [50]. Кроме того, у мышей с нулевым Sox11 и сниженной экспрессией Fgf9 наблюдается уменьшенная нижняя челюсть и CP, напоминающая CP, вызванную микрогнатией и неправильным положением языка в Pierre Robin Sequence (PRS) [51]. В недавнем исследовании повышенный уровень Fgf9 также вызывал дисплазию височно-нижнечелюстного сустава (TMJ), нарушая пространственную координацию между опусканием языка и подъемом нёбных половин, тем самым усугубляя формирование CP. Дисплазия височно-нижнечелюстного (TMJ) сустава ограничивает задний размер нижней челюсти и увеличивает нагрузку на заднюю часть нёба, тем самым повышая вероятность формирования расщелины. Эти данные свидетельствуют о том, что дисплазия височно-нижнечелюстного сустава, которая также сопровождает CP при таких синдромах у человека, как ахондроплазия и синдром Muenke может способствовать формированию CP , даже не уменьшая длину нижней челюсти [52].
Экспрессия LIM-гомеобоксных генов Lhx6 и Lhx8 негативно регулирует пролиферацию верхнечелюстной дуги и нёбной мезенхимы путем репрессии факторов транскрипции семейства FOX и ингибитора клеточного цикла Cdkn1c (p57Kip2) (рис. 3E) [53]. Поддержание митохондриального гомеостаза с помощью Lhx6 осуществляется через PINK1/Parkin-опосредованную митофагию и сигнальный путь MAPK. Транскрипционное снижение Lhx6 под действием RA нарушает поддержание митохондриального гомеостаза на транскрипционном уровне, что приводит к дефектам в пролиферации и миграции клеток HEPM и формированию CP [54]. Хотя известно, что молекулярная цепь Shh-Foxf1/2-Fgf18-Shh участвует в раннем развитии нёба (рис. 3E), неясно, влияет ли Lhx6/8 также на сигнальную сеть Shh и FGF во время формирования нёбной перегородки. Кроме того, трансформирующий фактор роста-β (Tgf-β) влияет на Shh-сигнализацию в нёбной мезенхиме, регулируя липидный обмен [55].
Сигнальный путь костного морфогенетического белка (Bmp) регулирует клеточную пролиферацию, клеточную дифференцировку и апоптоз, которые являются критическими этапами в морфогенезе лица [56,57]. BMP-путь может взаимодействовать с другими клеточными путями, такими как сигнальный путь Shh, который играет важную роль в развитии черепно-лицевой области [58] и взаимодействует с нёбной мезенхимой, где потеря Smo приводит к увеличению экспрессии Bmp4 и снижению уровня Bmp2 (рис. 3E) [48]. Сигнализация Shh способствует активности Bmp2 для стимуляции клеточной пролиферации в нёбной мезенхиме [59]. Роль Bmp2 заключается в развитии лицевых отростков в черепно-лицевом морфогенезе, а основная роль Bmp4 - в дифференцировке тканей, наряду с формированием лицевых отростков [60]. Хотя полная инактивация Bmp4 приводит к летальному исходу на ранних эмбриональных стадиях, его направленная делеция в мезенхиме верхнечелюстной кости и эпителии ротовой полости приводит к развитию CL , не затрагивая вторичное нёбо [56]. Избыточная экспрессия антагониста BMP Noggin в нёбной мезенхиме вызывает задержку роста нёба и к CP [61]. Это подчеркивает важную роль Bmp-сигнализации в нормальном нёбогенезе, а дисрегуляция этого процесса приводит к CL или CP [62]. Исследования показали, что Bmpr1a, рецептор Bmp I типа, необходим для формирования нёба (рис. 3F) [56]. Удаление Bmpr1a в мезенхиме верхнечелюстной кости и эпителии ротовой полости мышей вызывает CLP, тогда как его потеря только в эпителии ротовой полости не приводит к этому [63]. Это позволяет предположить, что сигнализация Bmpr1a в мезенхиме имеет решающее значение для палатогенеза. Условная делеция Bmpr1a в НКС приводит к тяжелым черепно-лицевым дефектам [64], в то время как его инактивация в нёбной мезенхиме приводит к ограничению CP в переднем направлении и к снижению клеточной пролиферации. Потеря Bmpr1a также нарушает экспрессию Shh, что указывает на то, что взаимодействие BMP-SHH регулирует рост нёба [65]. Кроме того, потеря антагониста BMP - Noggin - вызывает CP с повышенным апоптозом и сниженной клеточной пролиферацией [66], что подчеркивает необходимость жесткой регуляции Bmp-сигнализации во время развития нёба.
Сигнализация WNT имеет решающее значение для Pax9-опосредованного развития вторичного нёба [67-69] и регулирует пролиферацию, миграцию и дифференцировку клеток [70]. У Pax9-/- мышей снижение уровней Axin2 и β-катенина и повышение экспрессии Dkk2 (рис. 3F,G) нарушало WNT-сигнализацию, но фармакологическое ингибирование DKK частично восстанавливало морфологию нёба. Инактивация Sostdc1 восстанавливает WNT-сигнализацию и спасает от CP (рис. 3G) [67]. Патогенные варианты генов пути WNT, такие как Wnt3a, связаны с NSCLP [71]. Нарушения сигнализации WNT могут приводить к CL/P а также ассоциированы с другими заболеваниями [8], включая рак [72] и скелетные нарушения [73].
У мышей Pax9-/- сигналы EDA/EDAR ниже по течению от WNT-сигнализации снижены, но не являются существенными для палатогенеза [69]. Стимуляция агонистами EDAR in utero восстановила CP у этих мышей, при этом складки выглядели дезорганизованными и не влияли на экспрессию Bmp4, Msx1, Fgf10 или Osr2. Эти исследования показывают, что Pax9 действует через сигнальный путь WNT, регулируя антагонисты WNT в нёбной мезенхиме (рис. 3E). Однако необходимы дальнейшие исследования, чтобы понять, как Pax9 регулирует гены-мишени WNT.
3.3.2. Molecular Regulation and Regional Patterning Along the Anterior–Posterior Axis of Palatal Development
Развивающиеся нёбные половинки молекулярно и морфологически регионализованы вдоль оси A-P, где области передней части экспрессируют транскрипционные факторы, отличные от факторов задней части [74]. Msx1 и Shox2 необходимы для пролиферации мезенхимных клеток переднего нёба (рис. 3F), в то время как задний регион экспрессирует Meox2 и Tbx22 (рис. 3G). Msx1 через Bmp4 в мезенхиме регулирует экспрессию Shh в переднем нёбном эпителии (рис. 3F) [59], тогда как Mn1 и Barx1 экспрессируются больше в задней части (рис. 3G) [75]. Msx1 поддерживает экспрессию Shh в переднем нёбном эпителии, регулируя Bmp4 в мезенхиме (Рисунок 3F), в то время как Mn1 и Barx1 экспрессируются в основном в задней части (Рисунок 3G). У мышей с нарушенными генами Msx1 или Mn1 наблюдался полный CP, но дефекты были специфичны для конкретного региона. У Msx1-/-мышей дефекты пролиферации наблюдаются только в передней части нёба, тогда как у Mn1-/-мышей дефекты роста наблюдаются в средней и задней части нёба [59,75]. У Shox2-/-мышей расщелина ограничена передним нёбом, тогда как заднее нёбо развивается нормально, что свидетельствует о роли Shox2 в расширении переднего нёба [76]. Напротив, у Tbx22-/- мышей наблюдается различная степень тяжести расщелины, от полной CP до SMCP, при этом Tbx22 действует ниже по течению Mn1 в процессе роста заднего нёбного отростка [75]. Экспрессия Msx1 и Shox2 в переднем нёбе регулируется BMP-сигнализацией, о чем свидетельствует снижение экспрессии у Wnt1-Cre; Bmpr1af-/- мышей [64]. В культурах нёбных эксплантатов экспрессия Msx1 специфически индуцировалась в мезенхиме переднего нёба под действием Bmp4 [74], в то время как экзогенный Bmp4 не стимулировал экспрессию Shox2. Однако передний нёбный эпителий был способен индуцировать экспрессию Shox2 в задней мезенхиме, что выявило отчетливые различия между эпителием и мезенхимой вдоль передне-задний (A-P) оси (рис. 3F) [76]. Кроме того, каноническая Wnt-сигнализация ограничена передней нёбной мезенхимой и зависит от Gpr177 для секреции Wnt. В частности, в передней мезенхиме высок уровень экспрессии Wnt5a. Он регулирует мезенхимную миграцию и удлинение нёбной пластинки, а его транскрипция контролируется Msx1 (рис. 3F) [77]. В частности, факторы транскрипции домена LIM вместе с кофактором Ldb1 были идентифицированы в росте и формировании нёба, а их химическая и генетическая инактивация приводит к эктопической экспрессии Wnt5a в задней мезенхиме (рис. 3F) [78]. Эти данные подчеркивают различные молекулярные механизмы, вовлеченные в формирование A-P паттерна
3.3.3. Regulatory Networks and Patterning Along the Mediolateral Axis
Сигнализация SHH играет ключевую роль в развитии нёба, поскольку ее нарушение снижает экспрессию в нёбной мезенхиме [48]. Экспрессия Osr2 зависит от Pax9, и у эмбрионов, лишенных как Osr2, так и Pax9, наблюдается CP, а также снижение экспрессии Fgf10 в нёбной мезенхиме (рис. 3E). Это говорит о важности Osr2 и Pax9 в развитии нёба и регуляции уровня Fgf10 [79]. Паттернинг вдоль медиолатеральной оси нёба имеет решающее значение для создания доменов экспрессии генов, которые обеспечивают правильный рост и слияние. Около E12 на латеральной стороне нёбных пластинок начинают формироваться нёбные бороздки, и экспрессия Shh ограничивается этой областью [45]. С цинковыми пальчиками факторы транскрипции Osr1 и Osr2 имеют градиентную экспрессию вдоль медиолатеральной оси развивающейся нёбной мезенхимы (рис. 3E). К E13.5 экспрессия Osr1 ограничивается латеральной стороной, тогда как Osr2 сильно экспрессируется в латеральной мезенхиме, сужаясь к медиальной стороне. Удаление Osr2 приводит к CP из-за снижения клеточной пролиферации на медиальной стороне и нарушения паттернинга. Osr2 частично компенсирует роль Osr1, о чем свидетельствует восстановление CP у Osr2-дефицитных мышей с кДНК Osr1 [80].
У Osr2-/-мышей наблюдалась повышенная экспрессия генов, связанных с остеогенезом, таких генов в как Mef2c, Sox6, Sp7, и нескольких лигандов BMP (Bmp3, Bmp5 и Bmp7), а также эктопическая экспрессия семафоринов класса 3 (Sema3a, Sema3d и Sema3e). Это подчеркивает роль Osr2 в подавлении пролиферации мезенхимных клеток и предотвращении преждевременного остеогенеза. Функция семафоринов в палатогенезе еще не определена [81]. Dlx5-зависимый транскрипционный путь регулирует медиолатеральный паттернинг и расширение нёба (рис. 3E). Dlx5 экспрессируется в медиальной мезенхиме нёбной полки совместно с Fgf7; экспрессия последнего гена резко снижена в нёбе эмбрионов-мутантов Dlx5. Это снижение экспрессии Fgf7 может вызвать экспрессию Shh в медиальном нёбном эпителии, поскольку экзогенный Fgf7 может подавлять экспрессию Shh в культурах нёбных эксплантов. Хотя нёбные половинки у Dlx5-дефицитных мышей приподняты и сросшиеся, ротовое нёбо значительно увеличено, а мягкое нёбо деформировано [2].
Интересно, что хотя у мышей с дефицитом Msx1 экспрессия Shh в передней части нёба снижена, комбинированные мутанты, у которых отсутствуют и Dlx5, и Msx1, экспрессируют Shh в медиальном эпителии, компенсируя дефекты клеточной пролиферации, вызванные Msx1 [49]. Это исследование выявило новый путь, вовлекающий Dlx5 и Fgf7 в медиолатеральный паттернинг и рост нёба. Однако, поскольку у Fgf7-дефицитных мышей не наблюдается явных дефектов нёба, другая сигнальная молекула может действовать ниже по течению от Dlx5 для модуляции экспрессии Shh [1].
3.3.4. Genetic Network Controlling Palatal Shelf Adhesion and Fusion
Одновременно с этим рост нёбных полок поддерживает развитие верхнечелюстного и нижнечелюстного отростков, однако это происходит только благодаря движению языка вниз и вперед. Это необходимо для поднятия нёбных половинок, которые затем соприкасаются и срастаются по средней линии (рис. 1 и рис. 4B,C) [2]. В процесс, способствующий сцеплению и слиянию половинок, вовлечено сложное взаимодействие сигнальных путей. Мезенхимная целостность в сросшемся нёбе нарушается из-за удаления мезенхимы между половинками (рис. 4F). Нарушение дифференцировки краевого эпителия средней линии, способности к адгезии и потеря мезенхимально-эпителиального перехода могут привести к CP. Мутации или дисфункции в таких генах, как Jag2, Fgf10, Irf6 и Grhl3, приводят к недостаточной адгезии или слиянию нёбных полок, что приводит к CP [37,82]. Отсутствие Jag2, лиганда Notch, вызывает CP у мышей Jag2z ΔDSL/Delta;DSL в основном из-за аномального прилегания нёбных полок к языку. Jag2 экспрессируется в эпителии ротовой полости и поддерживает клетки перидермы, которые необходимы для регуляции компетентности слияния (рис. 4D) [37]. У эмбрионов Fgf10-/- также наблюдалось снижение экспрессии Jag2 и дефекты слияния нёба и языка, что позволяет предположить, что Fgf10 регулирует развитие нёба прежде сигнала Jag2-Notch (рис. 4D). Мыши с функциональными мутациями интерферон-регулирующего фактора 6 (Irf6), вызванными нулевым гомологичным сплайсингом или точечными мутациями R84C, демонстрируют не-дифференцированный гиперпролиферативный эпидермис, что приводит к различным аномалиям развития, включая CP и неадекватное сцепление с ротовой полостью [82]. Irf6 взаимодействует с Jag2 для регуляции эпидермальной дифференцировки, что подтверждается серьезными дефектами у мышей Irf6R84C/+; Jag2ΔDSL/+ [83]. Этот фенотип был схож с тем, который наблюдался у мышей с гомозиготными аллелями Irf6 или Jag2, что подчеркивает важность этих генов в развитии нёба (рис. 4D). У отдельных мутантов экспрессия обоих генов не нарушена, что указывает на то, что Irf6 не регулирует экспрессию Jag2 напрямую (рис. 4D) [82]. У мышей, лишенных фактора транскрипции p63, наблюдается CP и не-дифференцированный эпидермис [1], при этом экспрессия Irf6 в нёбном эпителии снижена [84]. У мутантных мышей, p63+/-; Irf6R84C/+, также не происходит слияние нёбных половинок из-за неправильного поддержания клеток перидермы. p63 может положительно регулировать экспрессию Jag2 и Fgfr2, хотя его связь с путями Jag2-Notch и Fgf10-Fgfr2b в дифференцировке нёбного эпителия до конца не изучена [85]. Отсутствие Ikk-α или Tbx1 у эмбрионов мыши приводит к аномальным ротовым адгезиям между языком и нёбными полками, это указывает на то, что дифференцировка нёбного эпителия контролируется генетической сетью, включающей сигнальные пути Irf6, Jag2, p63, Ikk-a, Tbx1 и Fgf10-Fgfr2b (рис. 4D)
Удаление перидермы и исчезновение медиального края нёбной половинки - два важных события, определяющих, произойдет ли нёбное сращение, и не возникнут ли такие аномалии, как аномальные оральные спайки рта. Однако механизмы, контролирующие удаление перидермы и исчезновение срединного эпителиального шва (MES), нуждаются в определении. Существуют три доминирующие гипотезы о том, как исчезает MES [87,88]. Согласно одной из гипотез, это может быть связано с эпителиально-мезенхимальным переходом (EMT). EMT может позволить эпителию MES интегрироваться в мезенхиму не-CP. Для отслеживания судьбы клеток MES было проведено несколько in vivo линейных анализов с использованием эпителиально-ограниченных Cre-экспрессирующих трансгенных линий и репортерных линий ROSA26R. Например, исследование экспрессии lacZ у мышей ShhGFPCre или K14-Cre, скрещенных с репортерными мышами R26R, не выявило присутствия lacZ-экспрессирующих мезенхимных клеток. Таким образом, был сделан вывод, что EMT не является основным механизмом регрессии MES [87]. В другом исследовании, напротив, сообщалось об активности мезенхимной β-галактозидазы в эмбрионах K14-Cre; R26R до и во время регрессии MES [89], что может быть связано с различными уровнями Cre или паттернами экспрессии между различными линиями трансгенных мышей K14-Cre.
ESRP1 и его паралог ESRP2 - эпителиальные белки-регуляторы сплайсинга, которые ко-локализуются с Irf6 и функционируют в эмбриональном эпителии, регулируя черепно-лицевое развитие и EMT во время эмбриогенеза (Рисунок 4D) [90]. Функциональные исследования показали, что мутации Esrp1/2 приводят к дефектному сплайсингу пре-мРНК, что, в свою очередь, вызывает аберрантные изоформы CTNND1, приводящие к ослаблению целостности эпителия и аномалиям OFC. Кроме того, исследования выявили ESRP1/2-контролируемые изоформы CTNND1, которые регулируют эпителиальную адгезию и WNT-сигнализацию, что указывает на нарушение сплайсинга при черепно-лицевых аномалиях (рис. 4D) [91].
Апоптоз играет решающую роль в растворении медиального края нёбной полки во время нёбного слияния, позволяя мезенхиме соединиться. Обычные признаки апоптоза, включая TUNEL-позитивность и активную каспазу 3, обычно обнаруживаются в клетках MES во время этого процесса, при этом в этой области наблюдается очень мало пролиферирующих клеток [87,92]. Однако недавние исследования, изучающие роль гена Apaf1, который участвует в апоптозе, опосредованном каспазой 3, показали, что дефицит Apaf1 не влияет на нёбное слияние или растворение MES [93], что противоречит предыдущим исследованиям, в которых сообщалось о проблемах слияния у эмбрионов с дефицитом Apaf1 [92]. Это может быть связано с тем, что в большинстве предыдущих исследований оценка нёба была неполной. Хотя апоптоз является одним из основных механизмов разрушения MES необходимы дальнейшие исследования для уточнения молекулярного механизма слияния нёба, включая Tgf-β-сигнализацию. Среди них Tgf-β3, экспрессирующийся исключительно в эпителии медиального края (MEE), играет важную роль в разрушении MES (рис. 4E). Отсутствие Tgf-β3 у эмбриональных мышей приводит к неправильному срединному контакту между нёбными пластинками и сохранению MES [1].
Сигнализация Tgf-β необходима для нёбного слияния и активируется через димеры рецепторов I и II типа, что приводит к фосфорилированию R-Smads и транскрипционной регуляции. Smad2 имеет решающее значение для разрушения MES, а избыточная экспрессия Smad2 может частично восстановить слияние у мышей с дефицитом Tgf-β3. Однако делеция Smad4 не влияет на слияние, что позволяет предположить участие других путей, таких как путь p38 MAPK [94]. Сигнализация Tgf-β активирует Tak1, который работает независимо от Smad-пути, способствуя нёбному слиянию как через Smad, так и через p38 MAPK-зависимые механизмы [2]. Irf6 регулирует дифференцировку перидермы и активируется в перидерме и базальных клетках MEE перед слиянием (рис. 4E). Его отсутствие в мутантных эмбрионах приводило к неудачному слиянию, однако сверхэкспрессия Irf6 восстанавливала этот процесс. Irf6 снижает регуляцию p63 и повышает экспрессию p21, способствуя выходу из клеточного цикла и дегенерации MEE (рис. 4E) [95,96]. Tgf-β3 снижает уровень Jag2 в MEE, а блокирование Notch-сигнализации может частично восстановить нёбное слияние в Tgf-β3-дефицитных культурах [97]. Целостность перидермы полости рта поддерживается Jag2-Notch-сигнализацией [37]. Снижение экспрессии Jag2 в MEE является ключевым механизмом, с помощью которого Tgf-β3 нарушает функцию перидермы и способствует сцеплению нёбных пластинок. Бета-катенин (Ctnnb1) также играет роль в нёбном слиянии, регулируя экспрессию Tgf-β3 в MEE. Разрушение β-катенина эпителиальных клеток (Ctnnb1) приводит к уменьшению количества апоптотических клеток MES в MEE, потере экспрессии Tgf-β3, отказу от слияния нёбных полок и CP. Однако β-катенин может функционировать в адгезивных соединениях или в каноническом сигнальном пути Wnt [98], и точный механизм его участия в рассасывании MES требует дальнейших исследований.
Несколько транскрипционных факторов имеют решающее значение для нёбного слияния. Семейство Snail, включая Snai1 и Snai2, играет ключевую роль, так как слияние не происходит у комбинированных мутантов Snai1+/-; Snai2+/- наряду с уменьшением апоптоза MES (рис. 4E). Интересно, что хотя экспрессия Tgfβ-3 у этих мутантов не нарушена [99], экзогенный Tgfβ- 3 может индуцировать экспрессию Snai1 через Smad-независимый путь, что позволяет предположить, что факторы Snail могут действовать ниже по течению или параллельно с сигналом Tgfβ-3. Runx1 - это транскрипционный фактор, участвующий в развитии нёба, который экспрессируется во всей MEE во время слияния нёба (рис. 4E) [1]. Нарушение работы Runx1 приводит к передне-специфическому нарушению слияния нёбных пластинок и образованию расщелины между первичным и вторичным нёбом. Эта неудача связана с отдельной областью в передней части MEE, с меньшим окрашиванием TUNEL и уникальным поведением [42]. Напротив, транскрипционный фактор Meox2 имеет решающее значение для поддержания целостности заднего нёба после слияния. У эмбрионов Meox2-/- наблюдается расщепление заднего нёба после слияния [93].
Irf6 необходим для экспрессии Snai2 в клетках MEE, а нокдаун Snai2 замедляет слияние нёбных дужек в культуре эксплантов [100]. Обратный сигнал эфрина усиливает экспрессию Snai1 в клетках MEE и может частично спасти слияние в присутствии антител, блокирующих Tgf-β3, что указывает на сотрудничество между эфрином и сигналом Tgf-β3 в регуляции нёбного слияния [101]. Snai1 и Snai2, действуя ниже по течению от Tgf-β3, снижают уровень E-кадхерина, что может ослабить адгезию клеток MEE и перидермы, приводя к десквамации перидермы (рис. 4E). Учитывая критическую роль Irf6 в этих процессах, регуляторная область MCS-9.7 может выступать в качестве мутационного очага для редких и распространенных генетических вариаций. Эти вариации могут приводить или повышать риск развития различных форм OFC нарушая экспрессию Irf6 в перидерме или базальных слоях эпителия полости рта [102]. Tgf-β3 и Irf6 также индуцируют MMP13, которая участвует в деградации базальной мембраны в MEE. CEACAM1, экспрессирующийся в перидерме до слияния, также вовлечен в процесс нёбного слияния, поскольку у Ceacam1-/- эмбрионов наблюдается задержка слияния, в то время как его связь с сигналом Tgf-β3 неизвестна [103]. Необходимы дальнейшие исследования, чтобы определить, как взаимосвязаны десквамация, апоптоз и Tgf-β3-опосредованная гибель клеток перидермы. Сигнальный путь TGF-β играет важную роль в ряде биологических и клеточных процессов, включая регуляцию клеточного роста, иммунные реакции и эмбриональное развитие [104]. Сигнализация Shh модулирует липидный обмен в нёбной мезенхиме. В морфогенезе лица сигнализация Tgf-β необходима для слияния нёбных дужек через взаимодействие с другими сигнальными путями, такими как WNT, FGF и BMP. Tgf-β участвует в EMT, что является жизненно важным шагом для успешной миграции и слияния нёбных половинок [105]. Сигнальный путь Tgf-β включает несколько генов, и было показано, что патогенные варианты некоторых из них связаны с развитием OFC например варианты гена IRF6, ассоциированного с VWS [18], семейства генов SMAD, которые также перекрестно взаимодействуют с сигнальным путем BMP, и варианты в этих генах связаны с повышенным риском развития CL [105]. Генетический процесс развития нёба включает в себя как генетические, так и эпигенетические факторы, в том числе миРНК, которые регулируют экспрессию генов во время нёбного слияния.
3.4. Epigenetic Mechanisms Landscape in Palatogenesis: Molecular Dynamics and Developmental Regulation 3.4.1. Overall Epigenetic Modifications in Development
Эпигенетика относится к механизмам, которые изменяют экспрессию генов путем модификации структуры хроматина, а не изменения самой последовательности ДНК [106]. ДНК плотно обернута вокруг белков гистонов, образуя нуклеосомы, которые являются основными единицами хроматина [107]. Расположение хроматина определяет, насколько ДНК транскрипционно активна; Эпигенетические модификаторы, к которым относятся «писатели», «стиратели» и «читатели», регулируют структуру хроматина с помощью различных механизмов, таких как модификации ДНК и гистонов, крупные белковые комплексы и не-кодирующие РНК. Метилирование ДНК обычно происходит в CpG-островках вблизи промоторных областей, а модификации гистонов включают добавление химических меток к хвостам гистонов, влияющих на рекрутирование транскрипционного механизма. Белковые комплексы, такие как репрессивные комплексы polycomb и комплексы ремоделирования хроматина, изменяют архитектуру хроматина и доступность ДНК. Не-кодирующие РНК, включая миРНК и длинные не-кодирующие РНК, участвуют в сайленсинге генов и регуляции хроматина (рис. 5) [108]. Когда эти эпигенетические регуляторы нарушаются в результате мутаций, экспрессия генов может аберрантно активироваться или репрессироваться, что приводит к таким заболеваниям, как рак и нарушения, связанные с нервным гребнем. Эпигенетические модификации предоставляют дополнительную генетическую информацию, которая может передаваться по наследству из поколения в поколение. Метилирование ДНК и модификация гистонов у млекопитающих регулируют экспрессию генов и влияют на судьбу клеток во время развития [109]. Эти динамические изменения могут меняться в ходе таких процессов развития, как черепно-лицевое развитие и развитие нервной трубки, регенерация тканей и старение [110]. Метилирование ДНК и модификации гистонов также регулируют геномный импринтинг, который является источником происхождения определенных генов. На эпигенетические модификации влияют факторы окружающей среды, такие как фолаты и ретиноиды, которые способствуют изменению результатов развития, включая NSOFC CLP и CPO [111]. Эпигенетические модификации могут объяснить различия в распространенности OFC в разных популяциях, которые нельзя объяснить только генетическими различиями [112].
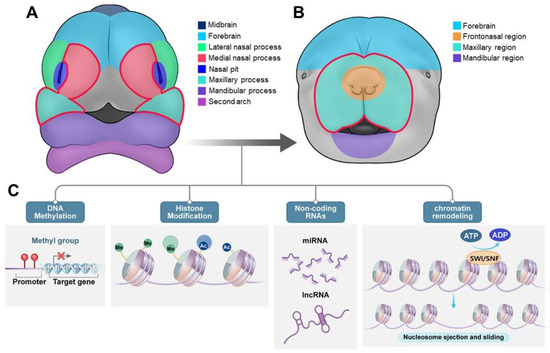 Figure 5. Epigenetic regulation during craniofacial development. (A,B) Schematic representation of early craniofacial development showing the morphological changes from initial facial prominences to their fusion. (A) The left panel shows the initial facial prominences, including midbrain, forebrain, lateral and medial nasal processes, nasal pit, maxillary and mandibular processes, and second arch. (B) The right panel shows the subsequent development of the frontonasal region, maxillary region, and the mandibular region. (C) Four major epigenetic mechanisms regulating craniofacial development: DNA methylation: addition of methyl groups to promoter regions controlling target gene expression. The red cross symbol means that the expression of the target gene is suppressed. Histone modification: post-translational modifications, including methylation (Me) and acetylation (Ac) of histone proteins. Non-coding RNAs: involvement of microRNAs (miRNA) and long non-coding RNAs (lncRNA) in gene regulation. Chromatin remodeling: ATP-dependent nucleosome ejection and sliding mediated by SWI/SNF complexes. Color code: dark blue—midbrain; light blue—forebrain; green—lateral nasal process; red—medial nasal process; navy blue—nasal pit; turquoise—maxillary process; purple—mandibular process and second arch; orange—frontonasal region; gray—other facial region behind maxillary/mandibular regions.
3.4.2. DNA Methylation Dynamics in Palatogenesis and Craniofacial Development
Метилирование ДНК включает в себя присоединение метильной группы к нуклеотидам цитозина в CpG-последовательностях [113], часто в CpG-островках в промоторных областях. Этот процесс рекрутирует транскрипционные репрессоры, которые подавляют экспрессию генов, блокируя факторы транскрипции (рис. 5) [114]. У млекопитающих и других позвоночных метилирование цитозина (C) в положении C5, приводящее к образованию 5-метилцитозина (5mC), широко признается в качестве единственной эпигенетической формы метилирования ДНК [8]. Метилирование аденина (A) у позвоночных остается спорным, в то время как о метилировании гуанина (G) и тимина (T) не сообщалось. Метилирование ДНК катализируется ДНК-метилтрансферазами (DNMTs), которые используют S-аденозилметионин (SAM) в качестве эксклюзивного донора метильных групп. Таким образом, активность DNMTs (гистон-модифицирующих ферментов) зависит от адекватного поступления SAM, который образуется в результате фолатного и метионинового циклов [112].
Считается, что метилирование способствует глушению генов, препятствуя связыванию механизмов транскрипции или активаторов с ДНК посредством пространственной интерференции. Кроме того, метил-CpG-связывающие белки, рекрутированные к 5-метилцитозину (5mC), могут активировать транскрипционные репрессоры, такие как деацетилазы гистонов [115]. Метилирование обычно происходит в цис-регуляторных элементах, в частности в промоторах и энхансерах, для контроля вариабельности экспрессии генов [116]. Промоторы, прилегающие к 5'-нетранслируемой области (5'-UTR), являются местом связывания транскрипционного механизма и запуска транскрипции. Энхансеры, расположенные рядом с промотором или в отдаленных областях, включая 3'-не-транслируемую область (3'-UTR), взаимодействуют с транскрипционными факторами, способствуя экспрессии генов. Они могут физически образовывать петли, взаимодействуя с промоторами и способствуя активации транскрипции [117]. Активность энхансеров высоко тканеспецифична и играет ключевую роль в пространственно-временной динамике экспрессии генов во время эмбрионального развития [118]. Хотя многие исследования были посвящены метилированию промоторов [116], последние данные свидетельствуют о том, что метилирование энхансеров и тел генов может играть равную или даже большую роль в регуляции экспрессии генов во время развития [119]. Метилирование происходит в определенных последовательностях оснований или мотивах и регулирует экспрессию генов в разных поколениях. Эпигенетическое метилирование обычно встречается в регионах, обогащенных 5'-CpG-3' мотивами, известными как CpG-островки [116]. Метилирование на островках CpG симметрично поддерживается на обеих линиях, что делает их наследуемым эпигенетическим маркером, не требующим de novo метилирования для восстановления. Промоторы тесно связаны с CpG-островками, в то время как энхансеры могут быть связаны с ними, а могут и не быть. Из-за корреляции между промоторами и CpG-островками первые исследования в основном фокусировались на метилировании промоторов как основном эпигенетическом контроле регуляции экспрессии генов. Другим важным эпигенетическим маркером является пятинуклеотидный мотив 5'-CCWGG-3' (где W может быть A или T), который подвергается метилированию внутри C. Метилирование 5'-CWGG-3' сохраняется в разных поколениях клеток млекопитающих, вероятно, благодаря тому, что метилтрансферазы распознают его как 5'-CCWGG-3'. Стабильное метилирование этого мотива опровергает существовавшее ранее представление о том, что только CpG-островки содержат C-основания, пригодные для трансгенерационного маркирования. Мотив 5'-CCWGG-3' имеет решающее значение для динамики метилирования энхансеров в орофациальном развитии [8] и привлекает все большее внимание.
Метилирование аденина, а именно превращение адениновых нуклеобаз в N6-метилдезоксиаденин (N6mA), является известной модификацией РНК, которая играет роль N6-метиладенозина (m6A) в процессинге РНК млекопитающих [120]. Однако их роль в метилировании ДНК млекопитающих остается спорной. Хотя N6mA является хорошо известной модификацией у прокариот и некоторых эукариот, таких как Caenorhabditis elegans, где она влияет на адаптацию митохондрий к стрессу [121], ее присутствие и функция в ДНК млекопитающих оспаривается. Последние исследования поставили под сомнение прежние утверждения о том, что N6mA является функциональным эпигенетическим маркером ДНК млекопитающих. Это позволяет предположить, что предполагаемые доказательства присутствия N6mA в ДНК млекопитающих могут быть вызваны загрязнением РНК или техническими проблемами [122]. N6mA может быть неправильно инкорпорирован в ДНК под действием полимераз при обработке рибо-N6mA, а не функционировать как настоящий эпигенетический маркер [123]. В РНК модификации N6mA хорошо изучены, особенно в контексте развития. Также обсуждалась потенциальная роль N6mA в РНК во время орофациального развития. Метилирование РНК, часто опосредованное генами семейства Nsun [112], высоко экспрессируется в эмбриональных тканях мыши, участвующих в черепно-лицевом развитии, что позволяет предположить, что модификации РНК могут играть определенную роль в этиологии OFCs.
Метилирование ДНК катализируется семейством ДНК-метилтрансфераз (DNMTs) [124], включая DNMT1, DNMT3А иDNMTs3В, которые играют важную роль в определении клеточной судьбы и тканевой спецификации, модулируя экспрессию генов. DNMT1 - это метилтрансфераза, которая поддерживает паттерны метилирования в процессе репликации и репарации ДНК. Функции метилирования de novo, с другой стороны, обеспечивают DNMT3A и DNMT3B, которые устанавливают новые метки метилирования в основном в промоторной области. Эти ферменты играют важную роль в регуляции экспрессии генов, особенно на ранних стадиях развития и в тканеспецифических GRNs. У мышей DNMT3а и DNMT3b высоко экспрессируются в не-дифференцированных эмбриональных стволовых клетках, и их экспрессия снижается по мере дифференцировки клеток [106]. DNMT3A репрессирует нейрональные гены, такие как Sox2 и Sox3, способствуя спецификации нервного гребня во время развития цыпленка [125]. DNMTb и метилтрансфераза G9a рыбок данио регулируют нейрогенез и формирование элементов черепно-лицевого скелета [126]. Исследования на эмбриональных стволовых клетках человека показали, что нокдаун DNMT3B ускоряет дифференцировку нейронов и нервного гребня за счет повышения регуляции генов спецификации нервного гребня, таких как PAX3, PAX7, FOXD3, SOX10 и SNAIL2 [127].
Исследования показали, что DNMT3B необходим для развития нервного гребня и черепно-лицевой области, однако условная потеря Dnmt3b в NCCs мыши приводит лишь к слабым дефектам миграции нервного гребня и не вызывает значительных черепно-лицевых фенотипов [128]. Это позволяет предположить, что DNMT3B может функционировать на более ранних этапах развития, чем считалось ранее, или может влиять на другие ткани, которые являются вторичными по отношению к развитию нервного гребня. Недавние исследования показали, что DNMT3B может функционировать без каталитического домена и выступать в качестве вторичного кофактора для поддержания ферментативной активности других DNMT, таких как DNMT3A [129]. Будущие исследования должны лучше изучить двойную роль DNMT3B, чтобы понять ее роль в развитии черепно-лицевой области и нервного гребня.
В нескольких исследованиях на мышиных моделях изучалась роль дифференциального метилирования генов в развитии орофациальной области, в частности в нёбном генезисе. Метилирование CpG в нёбе значительно выше на эмбриональный день E14.5, чем на E13.5 и E18.5 - критическое время, когда нёбные пластинки приподнимаются над языком, непосредственно перед формированием медиального эпителиального шва [8]. Для изучения метилирования ДНК в нёбе мышей от E12 до E14 был использован метод микрочипов. Они установили, что 73 % обнаруженных генов были метилированы, в основном в телах генов, а не в промоторах, причем 30 % метилирования происходило в CpG-островках [130]. Эти данные совпадают с результатами предыдущих исследований воздействия RA, в которых дифференциально метилированные регионы (DMR) располагались в интронных энхансерах генов, связанных с палатогенезом. Sox4, ключевой ген в развитии нёба, показал сниженную экспрессию в E13 и E14 из-за метилирования в CpG-бедной промоторной области. Sox4 играет роль в интеграции нескольких сигнальных путей, включая Tgf-β, Wnt/β-катенин, BMP, FGF и Hedgehog, которые регулируют слияние и расширение нёба [8].
Мутантные штаммы мышей также пролили свет на роль метилирования в развитии орофациальной области. Штамм A/WySn, у которого риск развития CL/P составляет 15-20 %, связан с эпистатическим взаимодействием между Clf1 (ретротранспозоном IAP на 3' конце Wnt9b), Clf2 и материнским эффектом. Clf2 подавляет IAP Clf1 через метилирование ДНК. Позднее Clf1 был идентифицирован как метастабильный эпиаллель со стохастическим метилированием во время эмбриогенеза, это указывает на то, что у некоторых особей отсутствует метилирование Clf1, что делает их уязвимыми к CL/P [131].
3.4.3. Epigenome-Wide Association Studies (EWAS) in Orofacial Clefts (OFCs)
В ходе EWAS были выявлены значительные различия в метилировании ДНК, связанные с OFC в частности с NSCLP. Например, исследование, проведенное в Великобритании, выявило дифференциально метилированные регионы в крови и ткани губы для всех подтипов расщелин, включая хорошо известные гены TBX1, COL11A2, HOXA2 и PDGFRA, и определило 250 новых локусов [11]. Более позднее исследование, проведенное в Бразилии, выявило 578 позиций варьирующего метилирования, связанного с NSCLP, которые были высоко обогащены для регуляторных областей, вовлеченных в черепно-лицевое развитие [132]. Было обнаружено, что Long interspersed nucleotide element-1 (LINE-1), маркер глобального метилирования ДНК, был дифференциально метилирован в NSOFC по сравнению с контролем [133]. Мутации I в гене 5,10-метилентетрагидрофолат-редуктазы (MTHFR), такие как c.C677T и cA1298C, снижают уровень метилирования ДНК [9], тогда как в центре CL при мутации c.C677T наблюдается повышенный уровень метилирования LINE-1 [121]. Эти DMRs могут объяснить отсутствие наследственных закономерностей при CL. В условиях воздействия окружающей среды можно ожидать популяционно-специфических эпигенетических модификаций, и для понимания их эпигенетического вклада в этиологию CL необходимо всемирное исследование популяций с расщелинами.
В последнее десятилетие профилирование метилирования позволило выявить эпигенетические модификации в качестве ключевых игроков в этиологии OFC. Эти модификации особенно привлекательны как механизмы экологических причин OFC. Например, было показано, что материнское курение дифференциально метилирует гены, ранее ассоциированные с OFC у детей, включая MSX1, PDGFRA, GRHL3, ZIC2 и HOXA2 [134]. Другие исследования по профилированию метилирования выявили гены с переменным уровнем метилирования, которые могут влиять на частоту возникновения OFC. К ним относятся факторы транскрипции (LHX8, PRDM16, PBX1, GSC, VAX1 и MYC), факторы и модуляторы роста (WNT9B, BMP4, EPHB2, BICC1 и DHRS2), гены внеклеточного матрикса (CRISPLD2, NTN1 и CDH1) и миРНК (MIR140 и MIR300) [132]. Некоторые из этих генов, в том числе PRDM16, BHMT2 и WHSC1, кодируют белки, участвующие в активности метилтрансфераз [135]. В дальнейших исследованиях были изучены позиции переменного метилирования в различных подтипах OFC и выявлены сотни позиций переменного метилирования, отличающих CLP, CLO и CPO [11]. Эти результаты свидетельствуют о том, что профилирование метилирования ДНК является перспективным подходом, который может дать более детальное понимание этиологии OFC.
3.4.4. Impacts of Histone Modifications in Craniofacial Development
Модификации гистонов - химические изменения гистоновых белков - имеют сложную природу и играют важную роль в регуляции структуры хроматина и экспрессии генов, особенно в процессе развития (рис. 5). В частности, такие PTM, как метилирование, ацетилирование, деацетилирование, фосфорилирование, убиквитинирование и сумоилирование, динамически модулируют экспрессию генов путем уплотнения и расслабления хроматина [108], а значит, и доступность хроматина. Эти изменения тесно связаны с решениями о судьбе клеток во время развития NCC и регуляция этих изменений особенно важна [136]. Другим важным PTMs влияющим на доступность хроматина и экспрессию генов, является ацетилирование гистонов, которое регулируется ацетилтрансферазами гистонов (HAT) и деацетилазами гистонов (HDAC) (рис. 5).
Миграция и дифференцировка NCCs также зависят от функции HDAC, а мутации в HDAC1, HDAC2 и HDAC4 связаны с черепно-лицевыми дефектами [137]. Кроме того, NCCs зависят от специфических модификаций гистонов, таких как H3K4me1 и H3K27ac, в энхансерных областях, которые регулируют структуру хроматина и экспрессию генов во время развития [138]. Метилирование ДНК и модификации гистонов часто работают вместе, и такие факторы окружающей среды, как воздействие 2,3,7,8-тетрахлордибензо-п-диоксина (TCDD), могут нарушать ацетилирование гистонов, тем самым влияя на процессы развития, такие как формирование CP у мышей [139]. Мутации в генах гистон-модифицирующих ферментов часто ассоциируются с нарушениями развития, такими как OFC [16]. В совокупности функции таких ферментов, как гистоновые метилтрансферазы, деметилазы, HATs и HDACs, имеют решающее значение для правильного развития нервного гребня. Нарушение этих процессов может повлиять на пролиферацию, миграцию и дифференцировку NCCs что приводит к врожденным черепно-лицевым дефектам.
Histone H3K27me3 Demethylase KDM6A, KDM6B
Синдром Кабуки, вызванный мутациями в KDM6A или KMT2D, приводит к аномалиям развития. KDM6A, Х-сцепленная H3K27 деметилаза, ассоциируется с замедленным ростом и CP, что наблюдается у одного пациента с гаплонедостаточностью [140]. Исследования на рыбках данио подтвердили, что снижение экспрессии kdm6a приводит к черепно-лицевым дефектам, что подтверждает ее роль в формировании CP [141]. Условный нокаут Kdm6a в NCCs с помощью Wnt1-Cre у мышей выявил половые эффекты, причем у самок наблюдались более тяжелые фенотипы, включая CP. Самцы могут компенсировать потерю Kdm6a за счет Y-связанного гомолога без деметилазной активности. Несмотря на важность Kdm6a в развитии нервного гребня, изменений в триметилировании H3K27 или H3K4 не наблюдалось, что позволяет предположить, что Kdm6a регулирует развитие через механизмы, независимые от его деметилазной активности [142].
Kdm6b является критическим игроком в развитии черепного нервного гребня, и потеря Kdm6b нарушает активность, опосредованную P53-путем, что приводит к полной CP, а также к дефектам клеточной пролиферации и дифференцировки у мышей. Kdm6b и Ezh2 антагонистически контролируют активность H3K27me3 в промоторе Trp53 в краниальных NCCs. Что еще более важно, в отсутствие Kdm6b транскрипционный фактор Tfdp1, который обычно связывается с промотором Trp53, не активировал экспрессию Trp53 в нёбных мезенхимных клетках. Более того, экспрессия Trp53 в таких клетках не может быть компенсирована высоко гомологичной гистоновой деметилазой Kdm6a [143].
Histone H3 Lysine 4 Methyltransferase KMT2D
Мутации в KMT2D, аналогичные мутациям в KDM6A, связаны с синдромом Кабуки. Исследование на модели Xenopus показало, что нокдаун kmt2d влияет на рассеивание NCCs, но не на другие виды миграционного поведения, что подтверждает роль KMT2D в метилировании H3K4 [144]. В этом исследовании в качестве мишени был определен sema3f, ген, необходимый для миграции краниальных NCCs , а его избыточная экспрессия частично восстановила этот фенотип. В отличие от этого, условный нокаут (cKO) Kmt2d у мыши показал полностью пенетрантный CP, но не повлиял на миграцию NCCs. Это наблюдение согласуется с тем, что наблюдается у пациентов с синдромом Кабуки [145]. Эта расщелина неба связана с аномальной экспрессией членов внеклеточного матрикса. Мутации Kmt2d могут влиять на другие цис-гены, а их последующие эффекты включают дисфункцию RAP1A и нарушение активации RAS/MAPK [146]. Ингибиторы MAPK-сигнализации, такие как desmethyl-dabrafenib, способны предотвратить структурные дефекты в ходе эмбриогенеза в модели синдрома Кабуки у рыбок данио, не оказывая при этом токсического воздействия [147], и могут представлять собой будущую терапевтическую стратегию.
Histone-Lysine Demethylase PHF8
Мутации в гистоновых деметилазах, таких как PHF8, которая деметилирует H4K20 и H3K9, могут приводить к тяжелым нарушениям развития. Мутации PHF8 связаны с CL/P и Х-сцепленной умственной отсталостью [148]. Деметилазная активность PHF8 необходима для транскрипции рибосомальной РНК (рРНК) [149] и дифференцировки нейронов [150]. Его каталитический домен, 2OG-оксигеназа, указывает на связь между гипоксией и OFC особенно у детей матерей, подвергавшихся воздействию табачного дыма во время зачатия [151]. У рыбок данио phf8 регулирует экспрессию msx1, которая связана с закладкой нервного ствола и черепно-лицевым развитием [150]. Избыточная экспрессия Phf8 у мышей также способствует регенерации костей, что указывает на потенциальное терапевтическое применение для лечения черепно-лицевых дефектов через регуляцию специального белка 2 (SATB2), связывающего богатые АТ-последовательности [152].
Histone-Lysine N-Methyltransferase MECOM (PRDM3)
N-метилтрансфераза MECOM (PRDM3) регулирует метилирование остатков H3K4 и H3K9. Во время развития рыбок данио prdm3 высоко экспрессируется в фарингеальных дугах [153]. Нокдаун морфолино приводит к дефектам нейронов и стеллатов и снижению экспрессии NCC-маркеров dlx2a и barx1. В другом исследовании схожие эффекты нокдауна Prdm3 наблюдались у развивающихся рыбок данио: снижалось метилирование H3K4 и H3K9 [154]. Условный нокаут Prdm3 с помощью Sox2-Cre у развивающихся мышей приводит к летальности в середине беременности.
Histone-Lysine N-Methyltransferase PRDM16
Prdm16 имеет схожие функции с prdm3 у рыбок данио, поскольку ее нокдаун снижает экспрессию маркеров нервного гребня dlx2a и barx1, а также снижает метилирование H3K4 и H3K9 [154]. Prdm16 имеет решающее значение для палатогенеза у мышей, как показали исследования потери функции с помощью мутагенеза, РНК-интерференции и устранения генов [155]. Во время развития нёба Prdm16 регулирует несколько целевых генов, участвующих в миогенезе, хондрогенезе и остеогенезе [156], а его потеря нарушает экспрессию Tgf-β и Bmp сигнальных путей. Условные нокауты выявили его роль в метилировании H3K9, но не H3K4 [154], и он также может регулировать развитие орофациальной зоны через транскрипционные факторы Smad [8].
Arginine Methyltransferase PRMT1
PRMT1 кодирует аргинин-метилтрансферазу, ответственную за модификацию H4R3me2a, и регулирует более 85 % активности метилирования аргинина, часть из которых направлена на негистоновые белки [157]. Условный нокаут Prmt1 в NCCs с помощью Wnt1-Cre приводит к черепно-лицевым порокам, включая CP, сходным с дефектами, наблюдаемыми у Msx1-нулевых мышей [158]. сКО Prmt1 снижает экспрессию Msx1 в критических черепно-лицевых областях на эмбриональный день 12,5. Последующее исследование выявило нарушение BMP-сигнализации у нокаутов, связанное с метилированием PRMT1 Smad6, ингибитора BMP. Снижение уровня H4R3me2a позволяет предположить, что роль PRMT1 в развитии также связана с модификацией гистонов [159].
Histone Methyltransferase WHSC1
WHSC1 - это ген метилтрансферазы, связанный с WHS, который экспрессируется как в эпителии, так и в мезенхиме во время развития нёба мыши. Его экспрессия снижалась, когда беременных мышей лечили полностью транс-ретиноевой кислотой (ATRA) - препаратом, который, как известно, вызывает CP [160]. Исследователи предположили, что whsc1 играет роль в стимулировании клеточной пролиферации. В отдельном исследовании нокдаун whsc1 у Xenopus привел к уменьшению ширины лица и уменьшению средней зоны лица. Он уменьшал расстояние миграции и общую площадь краниальных NCCs [161].
Histone Deacetylases HDAC3 and HDAC4
HDAC3 необходима для развития мыши, а условный нокаут в Histone Deacetylases HDAC3 and HDAC4 приводит к черепно-лицевым дефектам, включая CP [162]. HDAC3 регулирует транскрипционные факторы Msx1, Msx2 и Bmp4. При условном нокауте экспрессия этих генов увеличивается, в то время как пролиферация клеток снижается, а апоптоз увеличивается в возрасте E12.5. Ацетилирование гистонов, вероятно, важно для баланса экспрессии генов в NCCs .
HDAC4, гистоновая деацетилаза II класса, играет роль в остеогенезе, взаимодействуя с MEF2 и регулируя эндохондральное окостенение [8]. Во время развития рыбок данио hdac4 экспрессируется в премиграционных и мигрирующих краниальных NCCs. Нокдаун hdac4 приводит к уменьшению или отсутствию краниальных NCCs , что приводит к нёбным дефектам, таким как укороченные, расщепленные или отсутствующие ethmoid пластинки [163].
Histone Acetyltransferase KAT6A
Микроделеция или мутация TBX1 у людей вызывает синдром микроделеции 22q11.2, который включает такие симптомы, как SMCP, пороки сердца и дисфункция тимической железы [164]. У мышей делеция ацетилтрансферазы KAT6A, которая регулирует экспрессию Tbx1, частично имитировала синдром микроделеции 22q11.2, включая SMCP. Исследование показало, что дополнительная копия Tbx1 не устраняет дефекты нёба, вызванные дефицитом Kat6a, что позволяет предположить, что либо требуется более высокая доза Tbx1, либо Kat6a влияет на другие гены, помимо Tbx1.
3.4.5. Non-Coding RNAs in Craniofacial Development and Orofacial Clefts
миРНК - это небольшие (~22 нуклеотида) не-кодирующие молекулы РНК, которые регулируют различные процессы развития. миРНК обычно связываются с 3' нетранслируемыми областями (3'UTR) целевых мРНК благодаря комплементарности между начальной последовательностью миРНК и элементом ответа миРНК (MRE) [14]. Полное связывание приводит к деградации мРНК, а частичное - к подавлению транскрипции. миРНК могут регулировать несколько мРНК, а одна мРНК может быть мишенью для нескольких миРНК. Помимо сайленсинга генов, миРНК могут активировать транскрипцию, повышать экспрессию белков и воздействовать на митохондриальные транскрипты [14]. миРНК играют важную роль в эмбриональных тканях орофациальной области, воздействуя на гены, участвующие в клеточной пролиферации, апоптозе, дифференциации, клеточной адгезии и EMT (табл. 3) [165].
Формирование черепно-лицевых структур начинается с миграции и паттернинга NCCs. Dicer, фермент, необходимый для процессинга миРНК, критически важен для выживания постмиграционных NCCs, хотя он не требуется для их первоначальной миграции в лицевые примордии [166]. У нокаутных мышей, специфичных для NCCs наблюдаются различные дефекты черепно-лицевой области, в том числе микроцефалия, гипоплазия лица и CP [166]. Однако в моделях с нокаутом Dicer развитие нёба приостанавливается, поскольку скелетные структуры, образующиеся из NCC, остаются незрелыми или отсутствуют [166]. Большинство этих дефектов являются следствием обширного апоптоза, индуцированного производными NCCs и связаны с дефектом MAPK/ERK-сигнализации [166]. Функциональный анализ показал, что miR-21 и miR-181a репрессируют Sprouty2, негативный регулятор сигнального пути MAPK/ERK, необходимого для клеточной пролиферации, дифференцировки и апоптоза [167]. Из-за аномального паттернинга NCC нарушение Dicer у мышей приводит к серьезным дефектам верхнечелюстного, нижнечелюстного и фронтоназального отростков. miR-452 играет ключевую роль в регуляции EMT и паттернинга NCC, нацеливаясь на Wnt5a [168]. Потеря miR-452 увеличивает экспрессию Wnt5a и снижает сигналы Shh и Fgf8, тем самым уменьшая экспрессию Dlx2, ключевого регулятора паттернинга NCC в первой фарингеальной дуге [130]. Недавно miR-149 был вовлечен в этиологию несиндромальной расщелины губы с нёбом или без него благодаря своей роли в миграции клеток нервного гребня человека (hNCCs), полученных из индуцированных плюрипотентных стволовых клеток человека (рис. 6). С помощью 3' RNA-Seq в hNCC, избыточно экспрессирующих miR-149, было выявлено 604 дифференциально экспрессированных гена по сравнению с необработанными клетками, что подчеркивает их участие в этом процессе [169]. В работе [169] [170] продемонстрировали, что CP моделирует экспрессию генов и миРНК с E10.5 до E14.5 в верхнечелюстных отростках для выявления пространственно-временных закономерностей экспрессии генов и миРНК. МиР-325-3p и миР-384-5p, которые репрессировали гены, связанные с расщелиной, Adamts3, Runx2, Fgfr2, Acvr1 и Edn2, а их экспрессия увеличивалась с течением времени. Напротив, miR-218-5p и miR-338-5p подавляли связанные с расщелиной гены Pbx2, Ermp1, Snai1, Tbx2 и Bmi1, а их экспрессия снижалась с течением времени. Полученные данные свидетельствуют о том, что эти миРНК-мимики значительно подавляли пролиферацию клеток мезенхимальных клеток нёба мыши и линии O9-1 черепных NCCs путем модуляции CP-ассоциированных генов, что подтверждает их регуляторную роль в патогенезе [170].
 Figure 6. Summary of the miRNAs and genes associated with cleft lip and/or palate (CL/P) in mice and human. The complex miRNA-mediated regulatory networks involved in cleft lip and/or palate (CL/P). Environmental factors also affect cleft palate development by modulating miRNA activity, and how their dysregulation contributes to cleft formation through cell proliferation defects, differentiation defects, and cell death. Red arrows; graph showing genes going up, blue arrows; graph showing genes going down. CL, cleft lip; CP, cleft palate.
В процессе палатогенеза и развития OFC специфические миРНК играют важнейшую роль в регуляции клеточной пролиферации (рис. 6). Избыточная экспрессия miR-133b, miR-374a-5p и miR-4680-3p подавляет пролиферацию клеток в мезенхимных клетках нёба человека (HEPM), вероятно, за счет снижения регуляции GCH1, PAX7, FGFR2 и ERBB2 [14,171]. Аналогично, miR-497-5p и miR-655-3p снижают пролиферацию фибробластов губ человека, воздействуя на множество генов, связанных с OFCs в исследованиях CL/P. miR-124-3p снижает пролиферацию клеток клеточной линии миеломы (MELM), понижая уровень Bmpr1a, Cdc42 и Tgfbr1 [172]. В частности, была обнаружена сильная экспрессия miR-124-3p в верхнечелюстном отростке, особенно в возрасте E13.5. Кроме того, консервативная регуляторная подсеть человека и мыши, включающая пять транскрипционных факторов, включая GLI2, PAX3, PAX7, PAX9 и SATB2 [173]; три гена не-транскрипционных факторов, FGFR1, RARA и SUMO1; и пять миРНК, включая miR-27b, miR-133b, miR-205, miR-376b и miR-376c, контролируют пролиферацию мезенхимных клеток губы через следующие специфические гены-мишени: miR-27b - мишени PAX9 и RARA; miR-133b - мишени FGFR1, PAX7 и SUMO1; miR-205 - мишени PAX9 и RARA [174].
Кроме того, в работах. [171,175] выявили миРНК (miR-133b, miR-140-5p, miR-374a-5p, miR-381a-3p и miR-4680-3p), связанные с развитием CP у человека, с помощью систематических обзоров, биоинформационного анализа и анализа клеточной пролиферации в эмбриональных мезенхимных клетках нёба человека (HEPM) [171,175]. Используя данные пациентов с CP и HEPM клетками, в работе [176] было показано, что hsa-мезенхима является одним из основных факторов, способствующих развитию CP [176] продемонстрировали, что hsa-let-7c-5p и hsa-miR-193a-3p вовлечены в развитие CP, используя данные пациентов с CP и клеток HEPM. В работе [177] было показано, что гены hsa-let-7c-7c-3p и hsa-miR-193a-3p участвуют в развитии CP, используя данные больных CP и клеток HEPM. В работе [177] показано, что гены, предсказанные во всех трех базах данных (FUNRICH, MIRDB и Targetscan), рассматривались как потенциальные гены-мишени миРНК (104 гена-мишени miR-193a-5p, 512 генов-мишеней let-7c-5p). В частности, PIGA и TGFB2 были выбраны в качестве наиболее перспективных мишеней путем дальнейшего изучения трех баз данных (MGI, MalaCards и DECIPHER). Недавно в работе [178] было обнаружено, что Phenobarbital (PB) [178] специфически индуцирует экспрессию let-7c-5p, а специфический ингибитор let-7c-5p ослабляет вызванное PB подавление пролиферации клеток HEPM, это указывает на то, что let-7c-5p играет решающую роль в токсичности, вызванной PB. Let-7c-5p высоко экспрессируется в черепно-лицевых тканях эмбриональных мышей [178].
Исследования кластера mir-17-92 предоставили первые генетические доказательства того, что специфические миРНК функционально связаны с CL/P млекопитающих [179]. Кластер mir-17-92, состоящий из шести высоко консервативных миРНК (miR-17, miR-18a, miR-19a, miR-19b-1, miR-20a и miR-92a-1), относящихся к четырем семействам (miR-17, miR-18, miR-19 и miR-92), расположен на хромосоме 14 мыши (хромосома 13 у человека) [8]. Экспрессия mir-17-92 и двух его паралогов имеет сходный характер у эмбрионов мыши, снижаясь от E12 до E14 и концентрируясь в дистальных кончиках PS во время палатогенеза [165,179]. Прямыми мишенями miR-17-92 являются факторы T-box, такие как Tbx1 и Tbx3, которые содержат функциональные MRE в своих транскриптах. Эти гены повышены в miR-17-92 мутантных черепно-лицевых структурах, и исследования показали, что miR-17-92 напрямую репрессирует экспрессию Fgf10, который имеет решающее значение для правильного созревания нёбного эпителия [8,179]. Данные ChIP и ChIP-Seq продемонстрировали связывание AP-2a и Smad1/2/5 с хроматином miR-17-92, что позволяет предположить, что AP-2a и BMP-сигнализация регулируют экспрессию mir-17-92. Из этих исследований следует, что снижение регуляции специфических генов предшественников, таких как факторы T-box, с помощью miR-17-92 имеет решающее значение для нормального развития средней части лица [179].
Кластер miR-17-92 также нацелен на сигнальный путь Tgf-β в нёбной мезенхиме [8]. Снижение экспрессии miR-17-92 с E12-14 при одновременном увеличении экспрессии ключевых компонентов этого пути, таких как TGFBR2, SMAD2 и SMAD4, было очевидным в нёбных половинках. Люциферазный анализ в мезенхимных клетках нёба (PMCs) показал, что TGFBR2 является прямой мишенью miR-17 и miR-20a, в то время как SMAD2 и SMAD4 являются мишенями miR-18a [8]. Предполагается, что кластер mir-17-92 регулирует удлинение и повышение нёбной половинок путем регулирования TGF-β-индуцированного ингибирования пролиферации и синтеза коллагена. E2F1 напрямую связывается с промотором miR-17-92, способствуя его транскрипции. В нёбных мезенхимных клетках (НМК) обнаружена экспрессия E2F1 и E2F3 в тканях нёба в возрасте E12-14 лет, значительное подавление пролиферации клеток при нокдауне E2F1 и повышение регуляции кластера miR-17-92 при сверхэкспрессии E2F1 [180]. Чрезмерная клеточная пролиферация сдерживается, когда miR-17 и miR-20a связываются с 3-UTR E2F1, образуя таким образом петлю отрицательной обратной связи, которая помогает регулировать клеточный цикл. Нарушение регуляции этой петли отрицательной обратной связи может привести к формированию нёбной расщелины [180].
Слияние нёбных дужек происходит через разрешение медиального краевого шва (MES), что соответствует механизму «конвергенции и экструзии» на последних стадиях слияния нёбных дужек [181]. Он включает в себя создание временных эпителиальных мостиков над противоположными нёбными половинками, за которыми следует толстый эпителиальный слой, сходящийся в единый монослой благодаря клеточной интеркаляции и ороназальной транслокации эпителия MES [181]. В работе [181] [182] была идентифицирована miR-22-3p, регулятор сократимости актомиозина во время нёбного слияния. Ингибирование активности miR-22 в органных культурах нёба с помощью анти-miR-22 приводило к отказу от растворения MEE и удаления MES что подтверждает ключевую роль miR-22 в палатогенезе. Существует несколько потенциальных мРНК-мишеней miR-22 - это транскрипты, кодирующие две тяжелые цепи миозина (Myh9 и Myh10) и важные для сократимости актомиозина. Хотя функциональное взаимодействие между miR-22 и этими мишенями не было продемонстрировано в тканях нёба, мРНК Myh9 и Myh10 представляют собой функционально подтвержденные мишени для miR-22 в других биологических условиях [182].
Баланс транскрипционных факторов (TF) EMT важен для эффективного закрытия нёба. Сбалансированная регуляция эпителиальных и мезенхимных TF, таких как Grhl2 и Zeb1, необходима для закрытия нёба [183]. Считается, что Zeb1 отвечает за мезенхимную идентичность, тогда как Grhl2 и его мишени (например, Ovol1, Ovol2 и семейство миРНК miR-200) способствуют эпителиальной идентичности [183]. Результаты этого исследования показали, что Grhl2/miR-200 и Zeb1/Zeb2 противодействуют друг другу, и что Grhl2 трансактивирует семейство миР-200 миРНК для репрессии Zeb1/Zeb2 [183]. В этой связи было показано, что miR-200b нацелен на Smad2, Snail, Zeb1 и Zeb2, все они кодируют транскрипционные факторы, функционирующие как посредники сигнального пути Tgf-β. В ответ на Tgf-β активируется SMAD2/3. Он образует комплекс с SMAD4, который затем взаимодействует с ZEB1, ZEB2 или SNAIL, подавляя эпителиальные маркеры, стимулируя мезенхимные маркеры, вызывая миграцию и апоптоз [14,184]. Экспрессия miR-200b в MES постепенно снижается по мере слияния нёбных дужек, поскольку ингибирование клеточной пролиферации в MES необходимо для того, чтобы нёбные дужки могли слиться. Эти результаты указывают на то, что избыточная экспрессия miR-200b вызывает аномальный палатогенез в MES путем ингибирования Tgf-β-опосредованной экспрессии Smad2 и Snail [14,184].
Анализ MEE зародышей Tgf-β3-/-мышей в возрасте E13.5 показал, что экспрессия miR-206 в развивающемся нёбе была значительно снижена по сравнению с таковой у их собратьев дикого типа, что указывает на важную роль miR-206 в нёбном онтогенезе [14,185]. Профилирование экспрессии эмбриональных верхнечелюстных мезенхимных клеток мыши (MEMM), обработанных анти-миР-206, выявило значительные изменения в экспрессии ~230 генов, включая несколько членов суперсемейств Tgf-β и Wnt/β-катенин [185]. Аберрантная регуляция этого пути может вызывать различные нарушения, такие как OFC синдром Kallman, Crouzon и Apert [16].
Кроме того, экспрессия материнских миРНК влияет на риск развития расщелины. Например, избыточная экспрессия miR-152 была связана с черепно-лицевым дисморфизмом, а материнская циркулирующая miR-let7-3p может быть диагностическим биомаркером для NSOFC (табл. 3). SNPs в ферментах биогенеза миРНК или внутри миРНК-связывающих областей генов, связанных с расщелиной (например, FGF2, FGF9 и MSX1), были связаны с риском образования расщелины [174]. Исследование генетической ассоциации показало, что SNP (rs7205289:C > A), расположенный в предшественнике miR-140 (pre-mir-140), вносит вклад в несиндромальную предрасположенность к CP, влияя на процессинг miR-140 [177]. Минорный аллель А гена rs7205289, встречающийся с большей частотой у пациентов, ассоциирован с пониженной экспрессией miR-140-5p и повышенной экспрессией miR-140-3p [175]. Кроме того, этот аллель сохраняется у приматов и является функционально важным. Таким образом, дисрегуляция miR-140 может быть вовлечена в этиологию CP, что подтверждается генетическими данными. миРНК необходимы для правильной регуляции черепно-лицевого развития, и этот биомаркер подтверждается многочисленными дефектами черепно-лицевого развития, такими как CP и другие орофациальные дефекты, обусловленные дисрегуляцией миРНК [14].
Длинные не-кодирующие РНК (lncRNA) действуют как «губки» для миРНК, связываясь с определенными миРНК через MRE и снижая уровень миРНК [186]. Функция длинных не кодирующих РНК (lncRNAs) тесно связана с их субклеточной локализацией. В цитоплазме lncRNAs играют регуляторную роль, функционируя как миРНК-губки и участвуя в механизме конкурирующих эндогенных РНК (ceRNA). Кроме того, цитоплазматические lncRNAs могут взаимодействовать с РНК-связывающими белками (RBPs), оказывая тем самым биологическое действие [187]. Напротив, ядерная локализация lncRNAs позволяет им модулировать транскрипцию генов или предтранскрипционные процессы через взаимодействие с промоторными участками ДНК или транскрипционными факторами, называемое цис- и транс-регуляторными механизмами (табл. 3) [188,189].
Table 3. Regulatory roles of microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) in orofacial development.
SNP rs2262251 (G > C), расположенный в lncRNA RP11-462G12.2, оказался специфически связан с CL/P, но не с CP. Избыточная экспрессия аллеля G подавляет апоптоз и способствует пролиферации клеток HEK-293 и HEPM [14,190]. Исследования трансфекции с использованием репортеров люциферазы показали, что аллель C, но не G, специфически связывается с miR-744-5p. Трансфекция miR-744-5p в клетки HEPM снизила экспрессию lncRNA аллеля C, и это снижение было восстановлено с помощью ингибиторов miR-744-5p. И наоборот, избыточная экспрессия lncRNA снижала уровень miR-744-5p, подтверждая тем самым эффект сцепления lncRNA с С-аллелем [190]. Как miR-744-5p, так и IQSEC2 имели значительную обратную корреляцию и экспрессировались в тканях губ пациентов с CL/P. Таким образом, С-аллель lncRNA регулирует экспрессию IQSEC2 путем сдерживания miR-744-5p, усиления апоптоза и подавления пролиферации [190]. Было обнаружено, что LncRNA-NONMMUT100923.1 регулирует экспрессию Cdsn путем конкурентного связывания с miR-200a-3p в сети ceRNA во время палатогенеза, потенциально ингибируя адгезию эпителиальных клеток медиального края путем предотвращения дезинтеграции десмосомных соединений [188]. Например, в одном из исследований была отмечена потенциальная регуляторная роль NONMMUT004850.2/NONMMUT024276.2-миР-741-3p/miР-465b-5p-Prkar1a в нёбном слиянии во время развития CP [192]. Сложная регуляторная ассоциация с участием miR-483-3p, miR-4690-3p, miR-654-3p, miR-6515-5p, lncRNA RP11-731F5.2, lncRNA XIST, lncRNA RP11-591C20.9, RARA и SMPD1 была также выявлена в группах CL/P и CPO [38,193].
В некоторых работах, использовавших тот же набор данных lncRNA GSE183527, что и в нашем исследовании, было выявлено, что сети ceRNA (MALAT1-hsa-miR-1224-3p-SP1, MALAT1-hsa-miR-6734-5p/hsa-miR-1224-3p-WNT10A, NEAT1-hsa-miR-140-3p.1- CXCR4, NEAT1-hsa-miR-3129-5p/hsa-miR-199a-3p/hsa-miR-199b-3p-ZEB1 [194] и NEAT1-hsa-miR-130b-3p/hsa-miR-212-3p/hsa-miR-200b-3p-SMAD2) могут вносить вклад в этиологию NSOFC [188]. Сообщалось, что lncRNA TPT1-AS1 регулирует различные биологические процессы, включая клеточную пролиферацию, апоптоз, аутофагию, инвазию, миграцию и EMT, все они вовлечены в прогрессирование NSCL/P [188]. Была изучена потенциальная роль lncRNAs FENDRR и TPT1-AS1, а также мРНК EIF3H, RBBP6 и SRSF1 в развитии NSCL/P. Дальнейшее изучение механизмов регуляции этих lncRNA и их взаимодействия с другими факторами, такими как воздействие окружающей среды, может дать дополнительные сведения о развитии CP [187,188].
3.4.6. Epigenetic Regulation in Chromatin Organization and Craniofacial Development
Репрессивные комплексы Polycomb PRC1 и PRC2 являются важнейшими транскрипционными репрессорами, подавляющими экспрессию генов [195]. EZH2, каталитическая субъединица PRC2, подвергается метилированию H3K27. Условный нокаут Ezh2 в мышиных NCCs вызывает дерепрессию генов Hox, поддерживает NCCs в состоянии предифференцировки и нарушает остеохондро-прогениторные программы, которые влияют на формирование хряща и кости [196]. Мутации EZH2 также связаны с синдромом Weaver Overgrowth - генетическим заболеванием, вызывающим избыточный рост костей [197]. Ring1b/Rnf2, E3-убиквитин-лигаза в PRC1, регулирует дифференцировку черепно-лицевых хондроцитов. У мутантов ring1b рыбок данио наблюдается нарушение развития черепных хрящей и костей [198]. PHC1 и PHC2 являются ключевыми регуляторами экспрессии генов HOX и черепно-лицевого паттерна, и было показано, что генетические мутации, затрагивающие эти белки, лежат в основе синдрома CATCH-22, ассоциированного с фенотипами, подобными синдрому микроделеции 22q11.2, и NSCLP [199]. Белки ASXL (ASXL1/2/3), строительные компоненты комплекса PRC, имеют решающее значение для раннего развития нервного гребня. Мутации в ASXL вызывают такие нейроразвивающие заболевания, как синдромы Bohring–Opitz, Shashi–Pena и Bainbridge–Ropers, характеризующиеся черепно-лицевыми дефектами и задержкой развития [200].
АТФ-зависимые комплексы ремоделирования хроматина изменяют структуру хроматина АТФ-зависимым образом для контроля экспрессии генов, обеспечивая доступ к факторам транскрипции и механизмам, связанным с ДНК. Комплекс SWI/SNF, содержащий субъединицы ARID1A, ARID1B, BRG1 (SMARCA4), BRM, BAF155, BAF170, SMARCA2 (SNF5), SMARCB1 (INI1) и SMARCE1, является важным регулятором экспрессии генов. Мутации в этих факторах связаны с синдромальными пороками развития, такими как синдром Coffin–Siris, который проявляется в виде задержки в развитии, характерных черт лица и пороков развития пятого пальца или пальца ноги (рис. 5). У пациентов с синдромом Коффина-Сириса часто встречаются мутации, затрагивающие субъединицы комплекса SWI/SNF - ARID1A или ARID1B. У мышей потеря Arid1a в NCC приводит к тяжелым черепно-лицевым дефектам. В исследованиях на людях мутации ARID1B нарушают переключение между ARID1A и ARID1B во время развития нервного гребня, что приводит к нарушению дифференцировки [201]. Мутации Brg1 также связаны с синдромом Коффина-Сириса, при этом потеря Brg1 нарушает выживание и дифференцировку NCCs. Brg1 необходим для сохранения пула мультипотентных NCCs, и его потеря приводит к черепно-лицевым дефектам у рыбок данио и мышей [202]. Более того, потеря некоторых основных субъединиц SWI/SNF, таких как BAF155 и BAF170, приводит к нарушениям в выживании, миграции и дифференцировке NCCs что в свою очередь приводит к черепно-лицевым дефектам [203]. Другие субъединицы комплекса SWI/SNF, включая SMARCA2, SMARCB1 и SMARCE1, также играют аналогичную роль в спецификации нервного гребня и развитии черепно-лицевой области [204]. Другой ремодельер хроматина, CHD7, также играет важнейшую роль в развитии нервного гребня, а его мутации вызывают синдром CHARGE. CHD7 взаимодействует с комплексом PBAF, регулируя экспрессию и миграцию нервного гребня. На животных моделях, включая зебрафиш и мышей, показано, что мутации в Chd7 воспроизводят черепно-лицевые и сердечно-сосудистые дефекты, встречающиеся при синдроме CHARGE [30].
Помимо АТФ-зависимых комплексов ремоделирования хроматина, в черепно-лицевом развитии важны и другие эпигенетические регуляторы, такие как SATB2. SATB2 - это хроматиновый ридер, который связывается с областями прикрепления ядерного матрикса для активации транскрипции. У людей транслокации, затрагивающие хромосомный регион 2q32-q33, который включает SATB2, ассоциированы с CP [106]. Мутации в SATB2 связаны с SATB2-ассоциированными синдромами, характеризующимися умственной отсталостью, задержкой развития, черепно-лицевыми и зубными аномалиями [205]. У мышей мутации SATB2 приводят к черепно-лицевым аномалиям, напоминающим человеческие фенотипы, и дефектам дифференцировки остеобластов [106]. SATB2 регулирует скелетный паттернинг, контролируя гены Hox, а затем запускает остеогенез, регулируя такие гены, как Runx2 и ATF4, а также гены черепно-лицевого паттерна, включая Pax9, Msx1, Alx4 и Lhx7. Эти данные показывают, что SATB2 играет двойную роль в развитии нервного гребня: он способствует выживанию NCC на ранних стадиях, а затем управляет остеогенной дифференцировкой во время формирования черепно-лицевой структуры.
3.4.7. Environmental Influences on Epigenetics and Craniofacial Development
Воздействие факторов окружающей среды, особенно тех, которые происходят внутриутробно, связано с повышенным риском. К ним относятся курение табака, употребление алкоголя и/или использование ряда лекарственных препаратов, таких как бензодиазепины, кортикостероиды, антибиотики и противоэпилептические средства [206]. Кроме того, органические растворители и пестициды были связаны с профессиональным воздействием [207]. Предполагается, что взаимодействие генетической предрасположенности и факторов окружающей среды приводит к развитию CP (рис. 6) [206,208]. Недавнее исследование продемонстрировало повышенный риск развития OFC из-за высоких концентраций токсичных элементов в различных биологических матрицах. Однако были обнаружены взаимосвязи между повышением концентрации основных микроэлементов (ETEs) и снижением риска. Потребление матерью продуктов питания, содержащих ETEs, таких как цинк (Zn), селен (Se), медь (Cu), кобальт (Co) и молибден (Mo), связано с заметным снижением риска OFC [207]. Эти результаты означают, что воздействие токсичных элементов, как загрязняющих окружающую среду, так и пищу, способствует риску развития OFCs. Напротив, ETEs обратно ассоциированы с OFC что указывает на защитную роль в отношении OFC [207].
RA, производное, образующееся в результате метаболизма ретинола (витамина А), является важной сигнальной молекулой, необходимой для морфогенеза и дифференцировки во время эмбрионального развития, включая морфогенез черепно-лицевой области. Давно известно, что дефицит витамина А и его метаболитов приводит к врожденным черепно-лицевым дефектам, а избыток ретиноидов вызывает дефекты у грызунов, включая CL/P [16,209]. В исследованиях роли RA-сигнализации у грызунов использовались различные подходы, такие как направленный нокаут рецепторов ретиноевой кислоты (RARs) и ретиноидных X-рецепторов (RXRs), нокаут или ингибирование ферментов, участвующих в метаболизмеRA, и добавление экзогенного RA [206]. Циркулирующий ретинол связывается ретинол-связывающим белком (RBP) и поглощается клетками-мишенями через рецептор RBP, STRA6 [210]. Клеточный ретинол-связывающий белок (CRBP) способствует превращению ретинола в ретинальдегид под действием ферментов алкогольдегидрогеназы (ADH) или ретинолдегидрогеназы (RDH), который затем окисляется до RA под действием ретинальдегиддегидрогеназ (RALDH1-3) [211]. RA активирует RARs и RXRs, которые регулируют транскрипцию генов [206]. Мутации в RARs (α, γ) приводят к CP и расщелинам средней части лица, а у мышей с дефицитом Aldh1a3 снижена активность RA что приводит к летальной атрезии хоанального канала [212]. Все три гена Cyp26 (Cyp26a1, Cyp26b1 и Cyp26c1), которые метаболизируют RA, дисрегулируются у мутантов Tbx1 [213,214], что приводит к накоплению RA и снижению экспрессии ключевых регуляторов Bmp2 и Fgf10, в результате чего развивается CP [215]. RA негативно модулирует трансдукцию сигнала Tgf-β путем усиления экспрессии Smad7 и снижения активности Smad2, в то время как Tgf-β3 репрессирует сигнал RA. Два пути взаимно регулируют общие корепрессоры TGIF1, RA и TGF-β-сигнализации [206,216]. ATRA, одна из форм RA, также опосредует свои эффекты через Notch-сигнализацию в клетках нёба, управляя апоптозом и клеточной пролиферацией. В предыдущих исследованиях воздействие ATRA увеличивало экспрессию Notch1 в эпителиальных клетках и Notch2 в мезенхимных клетках, одновременно снижая уровень CyclinD1, что ингибирует пролиферацию [217]. Он также снижает экспрессию ключевых генов (Lhx8, Msx1 и Msx2) в эмбрионах цыплят, влияя на развитие орофациальной зоны [218]. Взаимное подавление RA и Wnt-сигнализации имеет решающее значение для черепно-лицевого развития [219]. У Wnt-дефицитных мышей наблюдается повышенная активность RA, при этом RA подавляет сигнализацию Wnt/β-катенин и нарушает регуляцию клеточного цикла [206]. RA также поддерживает экспрессию Shh и Fgf8, но избыток RA подавляет сигнализацию Shh и усиливает апоптоз в верхнечелюстной ткани мыши. Shh регулирует уровень RA через активацию гена Cyp26, что подчеркивает сложное взаимодействие между путями RA, Shh и Wnt в черепно-лицевом развитии [209,219]. Сигнализация Notch как в эпителии, так и в мезенхиме может быть изменена после воздействия ATRA, что приводит к изменению сигнализации p21. Например, воздействие АТРА в возрасте E10 увеличивало Notch2 и уменьшало CyclinD1 в мезенхимных клетках между E12,5-14,5, что подавляло пролиферацию [217]. В другом исследовании было обнаружено, что воздействие ATRA в возрасте E12 увеличивает экспрессию Notch1 в клетках MEE в возрасте E15, одновременно подавляя апоптоз нормального эпителия [220]. АТРА также снижает экспрессию Lhx8, Msx1 и Msx2 в верхней челюсти эмбрионов цыплят и генов, регулируемых Fgf и RA-сигнализацией [218]. ATRA может влиять на Lef1-опосредованный перидермальный переход EMT у мышей, снижая экспрессию Ambra1, в то время как Cyp26b1 необходим для правильного нёбного генеза и опускания языка [221]. В работе [221] [222] показано, что введение ATRA индуцирует изменения в миРНК у мышей с CP. CP у мышей, леченных RA, был связан с повышением уровня miR-181a-5p, miR-410-3p, miR-3960, miR-1224-5p, miR-3970, let-7e-5p, miR-1907, miR-124-3p и miR-4680-3p [185,222]. Далее, в работе [185,222] было показано, что miR-140-3p, miR-351-5p и miR-503-5p были снижены в мышиной модели RA-терапии (рис. 6). В клетках mouse embryonic palatal shelf (MEPS), обработанных ATRA, повышается уровень miR-124-3p, что приводит к подавлению целевых генов, таких как Axin1, Fst, Vcan и Zeb1 [175]. Кроме того, в предыдущем исследовании было показано, что miR-129-5p и miR-340-5p подавляют рост клеток первичных эмбриональных нёбных мезенхимных клеток мыши и клеток O9-1, полученных из нервного гребня. В частности, miR-129-5p нацелена на Sox5 и Trp53, а miR-340-5p регулирует экспрессию Chd7, Fign и Tgfbr1 для модуляции клеточного роста [223]. MiR-106a-5p значительно (~8,9 раза) повышался в тканях CP, вызванного ATRA, а избыточная экспрессия miR-106a-5p снижала экспрессию Tgfbr2 и изменяла уровни pSMAD2 и pSMAD3 [224]. Кроме того, miR-106a-5p сильно отрицательно коррелировала с метаболитами холестерина, что позволяет предположить, что miR-106a-5p может играть предполагаемую роль в метаболизме холестерина через Tgf-β сигнализацию в дефектном палатогенезе CP вызванном ATRA [224]. Таким образом, способность miR-106a-5p модулировать апоптоз может происходить через TGFβ/SMAD-путь [14,224]. Недавно была выявлена регуляторная сеть ceRNA, в которой LncRNA-NONMMUT100923.1 регулирует экспрессию Cdsn путем конкурентного связывания с эндогенной miR-200a-3p во время палатогенеза в мышиной модели, индуцированной ATRA [191]. Недавно секвенирование РНК ткани нёбной перегородки мыши (MEPS) показало, что лечение ATRA значительно повышает экспрессию miR-470-5p и снижает экспрессию Fgfr1 по сравнению с контролем. Результаты последовательно продемонстрировали, что miR-470-5p ингибирует EMT в эпителиальных клетках MEPS, в первую очередь подавляя экспрессию Fgfr1 [225].
В исследованиях, проведенных в Китае, Демократической Республике Конго и Мексике [206,226], сообщалось, что употребление алкоголя связано с повышенным риском развития OFC. Примечательно, что объединенное исследование показало повышенный риск OFC связанный с употреблением алкоголя во время беременности [206,227]. Точные механизмы, с помощью которых алкоголь повышает риск развития OFC до конца не изучены, однако было предложено несколько тератологических гипотез. К ним относятся антагонизм РА-сигнализации, изменение эпигенетики и окислительный стресс, когда этанол (EtOH) окисляется до токсичного промежуточного ацетальдегида (AcAL) [228]. Эти механизмы включают окисление EtOH в токсичный промежуточный продукт, AcAL, который в конечном итоге метаболизируется до ацетил-КоА под действие
В модели конкуренции RA-EtOH AcAL конкурирует с ретинальдегидом за фермент, синтезирующий RA. Исследования, проведенные на Xenopus, показали, что воздействие EtOH ингибирует синтезRA, препятствуя активности Aldh1a2, который превращает ретинальдегид в RA [229]. Кинетический анализ человеческого ALDH1A2 выявил предпочтение AcAL перед ретинальдегидом в качестве субстрата, что дало биохимическую основу для модели конкуренции RA-EtOH. В другом исследовании на рыбок данио было обнаружено, что черепно-лицевые пороки у мутантов pdgfra усугубляются EtOH и что pdgfra защищает от EtOH через путь PI3K-mTOR [230]. EtOH может изменять эпигенетическое состояние нейральных стволовых клеток, влияя на метилирование генов клеточного цикла и усугубляя пороки развития у генетических мутантов [231]. Кроме того, метаболизм EtOH в AcAL под действием CYP2E1 может привести к окислительному стрессу, апоптозу и активации сигнальных путей, что может нарушить развитие орофациальной зоны [232]. В мышиных эмбриональных стволовых клетках EtOH и AcAL способствуют дифференцировке через транскрипционную активность рецептора RA, RAR-γ, который индуцирует экспрессию целевых генов, таких как Hoxa1 и Cyp26a1 [233]. Таким образом, воздействие EtOH может изменить клеточный состав OFC, но для начала хорошо спланированных исследований знаний уже достаточно [206].
Диоксины и родственные им соединения представляют собой группу сходных соединений, состоящих из двух coplanar benzene rings, вызывающих различные токсические фенотипы с разными потенциалами [14] . Каждое соединение оценивается фактором токсического эквивалента (TEF), базируясь на его токсичности относительно TCDD, который является наиболее токcическим компонентом в этой группе. TCDD является прототипическим компонентом в исследованиях механизмов токичности, при этотм тератогенность служит чувствительным показателем его эффектов у экспериментальных животных. Воздействие TCDD нарушает экспрессию генов эпителиальных и мезенхимных тканей, нарушая многие стадии палотогенеза, включая рост и слияние [206] . Экспериментальные исследования показали, что введение TCDD беременным хомякам, мышам и крысам вызывает CP у их плодов [206]. Рецептор ариловых углеводородов (AhR), также известный как диоксиновый рецептор, является важнейшим лиганд-активируемым транскрипционным фактором для тератогенных эффектов TCDD [235]. AhR высокоактивен в эпителиальных клетках нёбных пластинок, а также обнаружен в костной и мышечной тканях нёба. У плодов, подвергшихся воздействию TCDD, MEE покрыта более тонким эпителиальным монослоем с меньшим количеством филоподий, что нарушает правильное слияние нёбных половинок, в отличие от контрольных плодов, у которых эпителиальный слой толще [236]. Уменьшение числа филоподий и нарушение слияния также наблюдалось в ex vivo культурах нёбных половинок, подвергшихся воздействию TCDD [237]. Примечательно, что добавление Tgf-β3 восстанавливало слияние этих культур, что подтверждает предположение о его недостаточности при CP, вызванном TCDD Однако экспрессия Tgf-β3 повышена в нёбе мышей, подвергшихся воздействию TCDD что указывает на необходимость использования различных экспериментальных систем [238]. Кроме того, воздействие TCDD снижает экспрессию нескольких важнейших факторов, участвующих в развитии черепно-лицевой области, включая FGFR1, Runx2, остеопонтин (OPN), MyoD и десмин. Он изменяет экспрессию E-кадхерина и Sox9, что может способствовать формированию CP [14,206]. Кроме того, TCDD нарушает сигнализацию факторов роста во время нёбогенеза через AhR-сигнализацию, в частности, снижая экспрессию TGF-a в эпителиальных клетках нёба человека [239]. Это позволяет предположить, что TCDD воздействует на путь эпидермального фактора роста (EGF), что подтверждается результатами нокаутных исследований, несмотря на кажущееся нелогичным снижение экспрессии лиганда [206]. В фетальных нёбных эпителиальных клетках человека (hFPECs) воздействие TCDD стимулирует фосфорилирование рецептора EGF, что приводит к фосфорилированию ERK/p38 и увеличению пролиферации клеток [240]. TCDD также способствует переходу клеточного цикла в фазы S и G2/M через путь PI3K/AKT [240]. Примечательно, что воздействие TCDD нарушает переход от эпителия к мезенхиме, снижая экспрессию эпителиальных маркеров, таких как E-кадхерин и кератин-14, и повышая экспрессию мезенхимных маркеров, таких как виментин и фибронектин. Это происходит за счет индукции Slug, индуктора EMT и репрессора E-кадхерина, который содержит DRE в своем промоторе, модулируемом AHR. Это позволяет предположить, что неадекватный EMT через Slug является ключевым механизмом в AHR-опосредованном CP [206,240]. Это согласуется с предыдущими данными о том, что Tgf-β3 защищает от CP после воздействия TCDD [241]. В работе [241] [242] показано, что функция TCDD в основном связана с метаболическими процессами внутриклеточных соединений, включая метаболические процессы клеточных ароматических соединений и метаболизм экзогенных лекарств под действием цитохрома Р450. Кроме того, было выдвинуто предположение, что сети ceRNA, circRNA_1781/miR-30c-1-3p/PKIB и XR_380026.2/miR-1249-3p/DNAH10 участвуют в развитии нёба, что позволяет предположить, что сети ceRNA circRNA_1781/miR-30c-1-3p/PKIB и XR_380026.2/miR-1249-3p/DNAH10 могут быть критическими для нёба, что создает основу для исследования CP [242]. Недавно было обнаружено повышение уровня миРНК (miR-214-3p, miR-296-5p и miR-33-5p) в группе, обработанной TCDD в то время как уровни миРНК (miR-92a-3p, miR-126a-3p и miR-411-5p) были значительно снижены. Примечательно, что анализ методом qRT-PCR подтвердил значительную разницу в экспрессии miR-214-3P. Эти данные подтверждают, что TCDD ингибирует пролиферацию и миграцию мезенхимальных клеток нёба через miR-214-3p, что предполагает его роль в TCDD-индуцированном CP у мышей [243].
Дексаметазон (DEX) - синтетический глюкокортикоид (ГК), используемый в клинической практике во многих случаях благодаря своим противовоспалительным и иммуносупрессивным свойствам. Эти эффекты опосредованы взаимодействием с различными сигнальными путями и молекулами, включая Toll-подобные рецепторы и митоген-активируемые протеинкиназы (рис. 6) [244]. Механизм действия GC включает диффузию внеклеточных GC в цитоплазму, где они связываются с цитозольным GC-рецептором. В отсутствие GC-GR образует комплекс с белками теплового шока (HSP70 и HSP90), FKBP52 и p23. Связывание GC приводит к диссоциации этого комплекса, и образовавшийся комплекс GC GR формирует димер. Активированный димер затем транслоцируется в ядро и связывается с элементами глюкокортикоидного ответа (GRE) в промоторной области генов-мишеней, активируя транскрипцию (трансактивацию) [244]. В качестве альтернативы активный комплекс GC-GR может напрямую связываться в виде мономера с NF-κB (p50/p65), без димеризации. Этот мономерный комплекс, связанный с NF-κB, впоследствии связывается с элементами ответа NF-κB для репрессии транскрипции. Несмотря на терапевтические преимущества, GC проявляют тератогенные и токсические эффекты. Например, риск CL/Р, преждевременных родов и низкой массы тела при рождении увеличивается в два-девять раз после приема пероральных или системных кортикостероидов во время беременности [174]. DEX может преодолевать гематоплацентарный барьер и связываться с цитоплазматическим GR, вызывая CP у мышей путем ингибирования клеточной пролиферации в нёбной мезенхиме [244]. Недавно анализ экспрессии миРНК в развивающихся нёбных пластинках мыши выявил отчетливые изменения между эмбриональными днями E13.5 и E14.5. Семейство miR-449 (включая miR-449a-3p, miR-449a-5p, miR-449b, miR-449c-3p и miR-449c-5p) показало повышенную экспрессию на E4.5, тогда как miR-19a-3p, miR-130a-3p, miR-301a-3p и miR-486b-5p - пониженную. Далее была изучена функциональная роль этих миРНК в клеточной пролиферации, которая показала, что избыточная экспрессия семейства miR-449 и miR-486b-5p подавляет клеточную пролиферацию в первичных эмбриональных нёбных мезенхимных клетках мыши и черепной линии NCC O9-1. Напротив, при ингибировании miR-130a-3p и miR-301a-3p наблюдался противоположный эффект; снижение экспрессии этих миРНК, а также снижало клеточную пролиферацию в тех же типах клеток [245]. Эти данные позволяют предположить, что miR-130a-3p играет важную роль в дексаметазон-индуцированном CP у мышей [174,245].
Метаболизм фолатов является важным этапом эмбриогенеза. Метаболизм фолатов включает в себя сложные механизмы регуляции и взаимодействия с метиониновым циклом. Поступающая с пищей фолиевая кислота подвергается метаболической переработке в тонком кишечнике и печени, сначала восстанавливаясь до дигидрофолата, а затем до тетрагидрофолата (ТГФ) под действием дигидрофолат-редуктазы (DHFR). Под действием серин-гидроксиметилтрансферазы (SHMT) витамин B6 необходим для образования 5,10-метилен-THF, а затем 5-метил-THF под действием метилентетрагидрофолат-редуктазы (MTHFR). 5-Метил-THF поступает в клетки-мишени через переносчики фолатов и превращается в THF с помощью метионин-синтетазы, используя витамин B12 в качестве кофактора. В ходе этой реакции Hcy превращается в метионин. Затем метионин превращается в S-аденозилметионин - основной донор метила при метилировании биомолекул, включающем модификации ДНК и гистонов. Отдав метильную группу, SAM превращается в SAH, который перерабатывается в гомоцистеин и завершает цикл метионина [206]. Систематический обзор показал обратную ассоциацию между приемом фолиевой кислоты и OFC [206]. Перекрестное исследование среди населения Калифорнии показало снижение распространенности как CL/P (ОР = 0,91, 95% DI: 0,82, 1,00), так и CPO (ОР = 0,81, 95% DI: 0,70, 0,93) после обязательного обогащения зерна фолиевой кислотой [246]. Недавний мета-анализ показал, что добавки фолиевой кислоты защищают только в период перед беременности (ОР = 0,64, 95% DI: 0,56, 0,74) по сравнению с беременностью (ОР = 0,90, 95% DI: 0,71, 1,14) [247]. Недавние исследования выявили взаимодействие генов и окружающей среды (G - E), влияющее на OFC и метаболизм фолатов в чилийской популяции. Полиморфизм в SHTM1, реже встречающийся в случаях CL/P, может защищать от CL/P путем снижения ферментативной активности и повышения уровня фолатов в клетках [248]. Фолат критически важен для метилирования ДНК, поскольку фолатный цикл обеспечивает метильные группы для SAM, молекулы, которая, как известно, метилирует ДНК и гистоны через DNMTs и гистоновые метилтрансферазы, соответственно [112]. Аналогичным образом, три интронных аллеля MTR, участвующих в метаболизме SAM, могут быть протективными [248]. Обычный полиморфизм MTHFR c.677C > T снижает ферментативную активность MTHFR и повышает риск CL/P, особенно в сочетании с низким потреблением фолиевой кислоты матерью [249]. Этот результат был подтвержден мета-анализом, объединившим 15 исследований; материнские мутации 677C > T обусловливают предрасположенность к CL/P, подтверждая, что материнский метаболизм фолатов играет важную роль в развитии OFC [249]. По некоторым теориям, фолаты способствуют правильному метилированию ДНК и гистонов во время развития [244], в то время как другие считают, что они способствуют пролиферации клеток [250]. В исследовании [251] была проверена гипотеза о том, что профилактика фолиевой кислотой является эпигенетической модификацией, в частности путем определения того, связаны ли изменения в метилировании ДНК с CL/P. Используя массив Illumina® Human Beadchip 450K (https://bioconductor.org/packages/devel/data/annotation/html/IlluminaHumanMethylation450kanno.ilmn12.hg19.html, дата обращения: 10 января 2025 г.), оценивали уровни метилирования геномной ДНК в архивных пятнах крови новорожденных в исследовании случай-контроль с участием 182 человек. Случаи CL/P продемонстрировали значительное эпигеномное гипометилирование по сравнению с контролем; 63% CpG-сайтов показали пониженное метилирование у новорожденных. Это соответствовало расовым подгруппам. В общей сложности 28 CpG-сайтов достигли эпигеномной значимости, и все они показали гипометилирование в аномальных случаях. Наиболее значимым регионом с дифференциальным метилированием, связанным с CL/P, был ген VTRNA2-1, который также был гипометилирован в случаях (FWER p = 0,014) [251]. Этот регион был ранее описан как реагирующий на питание метастабильный эпиаллель. Изменения метилирования, связанные с CL/P, как правило, были более выражены в предполагаемых метастабильных эпиаллельных регионах или вблизи них. Анализ обогащения наборов генов, связанных с CL/P, выявил чрезмерную представленность генов, участвующих в развитии нёба, таких как WNT9B, MIR140 и LHX8. Эти изменения метилирования ДНК могут частично объяснить механизм, с помощью которого OFC реагируют на уровень фолатов в организме матери [251]. Полиморфизмы в других генах, связанных с фолатами, также были связаны с OFC. Дефицит фолата нарушает репарацию ДНК из-за неправильной инкорпорации урацила и способствует нестабильности генома [206]. Фолат также влияет на процессы, зависящие от S-аденозилметионина (SAM), включая синтез полиаминов для клеточной пролиферации, дифференцировки, апоптоза и метилирования ДНК в отношении эпигенетической регуляции [206]. Дефицит фолатов может приводить к накоплению токсичного гомоцистеина, а гипергомоцистеинемия ассоциируется с OFC. Повышенный уровень гомоцистеина увеличивает количество асимметричного диметиларгинина, реактивных видов кислорода (ROS) и окислительного стресса [252]. Таким образом, фолаты оказывают свое защитное действие благодаря участию в росте и дифференцировке клеток и поддержании физиологического баланса.
Фенитоин также известен как тератоген. В период с 1993 по 2007 год примерно 1 % населения получали рецепты на противоэпилептические препараты, и примерно 20 % этих рецептов были на фенитоин (Mayne Pharma Pty Ltd, Mulgrave, Австралия). У женщин детородного возраста этот процент увеличивается примерно до 5 %, что подчеркивает важность этого общепризнанного тератогена. У женщин с эпилепсией, принимающих фенитоин самостоятельно или в сочетании с другими препаратами, в два-три раза повышается риск рождения детей с врожденными пороками развития [253]. Гидантоиновый синдром плода, связанный с воздействием фенитоина, включает ограничение роста, типичные черты лица, включая гипоплазию средней части лица, повышенный риск развития расщелины губы, аномалии конечностей, которые чаще всего состоят в гипоплазии дистальных фаланг с маленькими ногтями, и повышенную частоту пороков сердца. Воздействие фенитоина в первом триместре беременности повышает риск развития гипоплазии верхней челюсти. В одном из исследований распространенность этого заболевания составила 16,7 % [254]. Связь между гипоплазией верхнечелюстной кости и CL рассматривается как взаимосвязанная аномалия, возникающая вследствие недоразвития верхнечелюстного отростка. Фенитоин в тератогенных дозах снижает скорость раннего эмбрионального сердца беременных крыс и вызывает увеличение продолжительности эмбриональной гипоксии, что объясняет наблюдаемые данные о том, что гипоксия повышает, а гипероксия снижает частоту возникновения CL у чувствительных штаммов мышей. Предполагается, что механизм, лежащий в основе этого эффекта, заключается в ингибировании фенитоином калиевого канала HERG наряду с уменьшением натриевых и кальциевых каналов, что также может объяснить действие на эмбриональное сердце [255]. В исследованиях на крысиных моделях фетального гидантоинового синдрома в одном помете постоянно наблюдались как расщелина губы, так и гипоплазия верхнечелюстной кости, что позволяет предположить наличие континуума по степени тяжести [254]. Кроме того, в работе [254] [255] было обнаружено, что фенитоин, являющийся индуктором CL, снижает пролиферацию клеток через индукцию miR-196a-5p. В период от E10,5 до E12,5 экспрессия miR-196a-5p значительно снижается в MxPs и NPs,, а фенитоин повышает уровень miR-196a-5p и подавляет пролиферацию клеток MELM путем снижения уровня Pbx1, Pbx3 и Rpgrip1l [256]. В частности, лечение специфическим ингибитором miR-196a-5p восстановило пролиферацию клеток за счет нормализации экспрессии CL-ассоциированных генов в клетках, обработанных фенитоином [255]. miR-196a-5p подавляет пролиферацию клеток и способствует остеогенной дифференцировке стволовых клеток пупочного канатика (WJCMSC) и подавляет формирование кости в WJCMSC-листе трансплантированной кальварии крысы за счет подавления Serpinb2 [256]. Эти коллективные данные свидетельствуют о плейотропной роли miR-196a-5p в различных процессах развития, в том числе критических для формирования нёба. Недавно в работе [257] было показано, что фенитоин ингибирует пролиферацию клеток HEPM дозозависимым образом путем снижения уровня циклина-D1 и циклина-E. Обработка фенитоином повышает уровень miR-4680-3p и снижает уровень его целевых генов, ERBB2 и JADE1. Эти данные свидетельствуют о том, что фенитоин подавляет клеточную пролиферацию путем модуляции miR-4680-3p [257].
Материнское курение было признано фактором риска в мета-анализе 24 исследований типа «случай-контроль» и «когорта», опубликованном в 2004 году в Бюллетене Всемирной организации здравоохранения, где сообщалось об ассоциации между материнским курением и CL/P [RR = 1,34; 95% CI: 1,25, 1,44] или CPO (RR = 1,22; 95% CI: 1,10, 1,35) [258]. Десять лет спустя в 50-м юбилейном докладе Генерального хирурга США было заявлено о достаточном количестве доказательств причинно-следственной связи между активным курением матери и OFC [259]. У человека имеется два гена NAT, NAT1 и NAT2, каждый из которых имеет более двух десятков известных полиморфизмов, которые могут влиять на детоксикацию ариламинов и риск развития OFC. Исследование, проведенное в калифорнийской популяции, выявило два фетальных полиморфизма NAT1, которые в четыре раза повышали риск развития CL/P, если матери курили на ранних сроках беременности (по сравнению с младенцами референтного генотипа от некурящих матерей [260]). Известно, что дым содержит эндогенные соединения табачных растений, такие как никотин, продукты пиролиза и дополнительные химические вещества. Соответственно, существует несколько гипотетических механизмов, посредством которых табачный дым повышает риск развития OFC. Генетическая предрасположенность существенно влияет на риск развития OFC вызванных табачным дымом. Полиморфизмы в генах, кодирующих сигнальные лиганды развития, такие как TGF-α, TGFβ3 и BMP4, были связаны с OFC в связи с воздействием материнского дыма [206]. Варианты, влияющие на взаимодействие между TGF-α и курением (взаимодействие гена и среды, GxE), были обнаружены в нескольких популяциях [206,261]. TGF-β3 кодирует лиганд, имеющий решающее значение для сигнализации TGF-β, необходимой для развития губы и нёба. Полиморфизмы в TGFβ3, ассоциированные с курением, были связаны с CL/P, CPO и SMCP [262]. BMP4 является лигандом для BMP-сигнализации, имеет полиморфизмы, связанные с CL/P, и играет роль в слиянии медиального и латерального носовых отростков. Варианты BMP4 в сочетании с курением также ассоциированы с CL/P [263]. Табачный дым влияет на экспрессию генов, регулирующих клеточный цикл, восстановление ДНК и реакцию на окислительный стресс в тканях плода мыши [264]. Кроме того, табачный дым может вызывать опосредованную протеасомами деградацию белков, которые опосредуют метилирование ДНК в клетках первой бранхиальной дуги (BA1) [265]. Никотин является вазоконстриктором, который может нарушать сосудистую функцию матки и влиять на кровоток и доставку кислорода к плоду [266]. В табачном дыме могут содержаться некоторые тератогены, такие как полициклические ароматические углеводороды , диоксины, угарный газ, пестициды и тяжелые металлы, например кадмий [267]. Воздействие тяжелых металлов может вызывать OFC в моделях грызунов и, как полагают, действует тератогенно через индукцию окислительного стресса и возмущение окислительно-восстановительных сигнальных путей [268].
4. Conclusions
Этот всеобъемлющий обзор иллюстрирует сложное взаимодействие генетических, эпигенетических и экологических факторов риска в регуляции палатогенеза и этиологии CL/P Эти исследования также прояснили сложные молекулярные сети, связанные с важнейшими сигнальными путями, включая TGF-β, BMP, FGF, Wnt и SHH, а также эпигенетические механизмы (например, не-кодирующие РНК, миРНК и длинные не-кодирующие РНК), которые способствуют развитию черепно-лицевой области. Последние данные свидетельствуют о необходимости точной регуляции различных этапов развития, включая пролиферацию, миграцию, ди
Дополнительную сложность создают факторы окружающей среды, включая материнское табакокурение, воздействие алкоголя, дефицит фолатов и тератогены, такие как RA и TCDD. Эти нарушения приводят к дисбалансу между генетическими и экологическими факторами, инициирующими нормальное развитие, что приводит к различным врожденным порокам развития, включая CL/P Последние достижения в области изучения взаимодействия генов и окружающей среды и эпигенетической регуляции позволят пролить свет на будущие методики профилактики и лечения. Остающиеся пробелы должны быть устранены в будущих исследованиях. В частности, роль не-кодирующих РНК и ремоделирования хроматина в развитии черепно-лицевой области требует дальнейшего изучения. Кроме того, важно определить точные молекулярные мишени, чтобы перевести полученные результаты в клиническую плоскость. Данный обзор объединяет генетические, эпигенетические и экологические аспекты, чтобы создать основу для будущих междисциплинарных стратегий по снижению бремени OFC.
|