Пользователи: 
СЕРДЕЧНАЯ НЕДОСТАТОЧНОСТЬ
Роль кальциевых каналов L-типа
Molecular Mechanisms of L-Type Calcium Channel Dysregulation in Heart Failure Arbab Khalid 1,Abu-Bakr Ahmed 2ORCID,Randeep Gill et al.
 Int. J. Mol. Sci. 2025, 26(12), 5738; https://doi.org/10.3390/ijms26125738
| |
The L-type calcium channels (LTCCs) function as the main entry points that convert myocyte membrane depolarization into calcium transients, which drive every heartbeat. There is increasing evidence to show that maladaptive remodeling of these channels is the cause of heart failure with reduced ejection fraction (HFrEF) and heart failure with preserved ejection fraction (HFpEF). Recent experimental, translational, and clinical studies have improved our understanding of the roles LTCC expression, micro-domain trafficking, and post-translational control have in disrupting excitation–contraction coupling, provoking arrhythmias, and shaping phenotype specific hemodynamic compromise. We performed a systematic search of the PubMed and Google Scholar databases (2015–2025, English) and critically evaluated 17 eligible publications in an effort to organize the expanding body of work. This review combines existing data about LTCC density and T-tubule architecture with β-adrenergic and Ca2+/calmodulin-dependent protein kinase II (CaMKII) signaling and downstream sarcoplasmic reticulum crosstalk to explain how HFrEF presents with contractile insufficiency and how HFpEF shows diastolic calcium overload and stiffening. Additionally, we highlight the emerging therapeutic strategies aimed at restoring calcium homeostasis such as CaMKII inhibitors, ryanodine receptor type 2 (RyR2) stabilizers, and selective LTCC modulators without compromising systolic reserve. The review establishes LTCC dysregulation as a single mechanism that causes myocardial dysfunction while remaining specific to each phenotype, thus offering clinicians and researchers a complete reference for current concepts and future precision therapy approaches in heart failure.
Т-канальцы (T-tubules) — это структурные элементы кардиомиоцитов, которые играют важную роль в процессе сокращения сердца. Они обеспечивают синхронизацию высвобождения кальция и поддерживают эффективную работу сердца.
Основные особенности и функции Т-канальцев:
В нормальных условиях Т-канальцы образуют хорошо организованную сеть, которая обеспечивает точное выравнивание L-тип кальциевых каналов (LTCC) с рианодиновыми рецепторами (RyR2) на саркоплазматическом ретикулуме. Это необходимо для координированного высвобождения кальция, которое позволяет сердцу сокращаться эффективно.
При сердечной недостаточности с уменьшенной фракцией выброса (HFrEF) архитектура Т-канальцев нарушается: сеть становится разреженной и неорганизованной, что приводит к разделению LTCC и RyR2. Это нарушает кальциевую сигнализацию и ослабляет силу сокращения.
Mode-2 gating (стробирование)— это состояни е работы L-типа кальциевых каналов (LTCC), которое характеризуется удлинённым временем открытия и высокой вероятностью нахождения канала в открытом состоянии.
|
Кальциевые каналы L-типа (LTCC) важны для осуществления связи между возбуждением и сокращением сердца (EC). Во время каждого удара сердца деполяризация мембраны приводит к открытию LTCC, которые в основном состоят из Ca_v1.2 в миоцитах желудочков, обеспечивая небольшой приток Ca2+, что, в свою очередь, вызывает гораздо большее высвобождение Ca2+ из саркоплазматического ретикулума (SR), процесс, называемый кальциево-индуцированное высвобождение кальция.[1,2]. Этот запускаемый LTCC сигнал Ca2+ быстро повышает уровень цитозольного Ca2+, активируя миофиламенты и вызывая сокращение, а обратный захват Ca2+ в SR затем приводит к расслаблению [1,2]. Благодаря такой эффективной связи электрического возбуждения с механическим сокращением с помощью LTCC, их активность играет важную роль в поддержании сердечного выброса и ритма.
При сердечной недостаточности (HF ) нарушение регуляции внутриклеточной регуляции Ca2+ является основной характеристикой, вызывающей сократительную дисфункцию и аритмии [1,3,4]. Неадекватные изменения функции или экспрессии LTCC нарушают взаимодействие с EC, вызывая менее эффективные сокращения и повышая уязвимость к аритмогенным постдеполяризациям. В целом, HF обычно подразделяется на два фенотипа — сердечную недостаточность сосниженной фракцией выброса (HFpEF) с расширением желудочков и систолической дисфункцией и сердечную недостаточность с сохраненной фракцией выброса (HFrEF) с почти нормальным систолическим выбросом, но нарушенным диастолическим расслаблением [5]. Примечательно, что HFrEF и HFpEF демонстрируют различные процессы ремоделирования и реакции на лечение, что позволяет предположить, что они имеют разные молекулярные механизмы [6,7]. Важно понимать, как изменяется регуляция LTCC при обоих подтипах HF , поскольку дисфункция LTCC способствует снижению сократительной способности и аритмогенезу при HF [3,8].
Этот обзор был оcнован на результатах поиска литературы в этой области с использованием PubMed (https://pubmed.ncbi.nlm.nih.gov, дата обращения - 15 апреля 2025 г.) и Google Scholar (https://scholar.google.com , дата обращения 15 апреля 2025 г.) базы данных для опубликованных статей за период с 2015 по 2025 год, посвященных молекулярным изменениям в регуляции LTCC при HFrEF и HFpEF. Мы подчеркиваем, что HFrEF обычно ассоциируется со сниженной экспрессией/функцией LTCC, измененными посттрансляционными модификациями и “расцеплением” связи EC, в то время как HFpEF, как правило, демонстрирует сохраненную экспрессию LTCC с неадаптивными сигнальными изменениями и диастолической дисфункцией. Новые методы лечения HF направлены на нормализацию нарушенной регуляции Ca2+ путем подавления гиперактивности CaMKII (с использованием таких пептидов, как AIP, KN 93 или новых низкомолекулярных ингибиторов [9]), стабилизацию каналов рианодиновых рецепторов типа 2 (RyR2) (Rycals, такие как ARM210, предотвращают диастолическую утечку Ca2+ [10])., и избирательно модулирующие Ca2+-токи L-типа (например, усиливающие ингибирующие взаимодействия Rad–LTCC или блокирующие повторно экспрессируемые каналы Cav1.3 [11]).
3. Discussion
3.1. LTCC Dysregulation in HFrEF
Сердечная недостаточность сосниженной фракцией выброса классически ассоциируется с ослаблением связи EC и снижением систолических переходных процессов Са2+ [12,13,14]. Отличительной чертой HFrEF является снижение экспрессии и функции LTCC в желудочковых миоцитах [15]. Исследования на поврежденных сердцах человека и моделях животных показывают, что пиковая плотность тока Са2+ L-типа снижается, часто на ~ 30-40% по сравнению с нормой, из-за потери LTCC в мембране и/или изменения стробирования каналов [4,16]. Это снижение LTCC способствует уменьшению переходных процессов Ca2+ и слабости сократительной способности при HFrEF [1-4]. Поврежденные желудочковые миоциты человека демонстрируют значительно более низкий ток LTCC и связывающую способность, которые могут частично восстановиться после механической разгрузки (поддержка устройства LV assist) [16]. Аналогичным образом, хроническая дилатация и повышенное напряжение стенки в моделях HFrEF приводят к снижению тока Ca2+ L-типа в т-канальцах и отсоединению от высвобождения SR Ca2+ [4,17,18].
Наряду с уменьшенным числом каналов, HFrEF характеризуется посттрансляционными модификациями - в первую очередь фосфорилированием α— и β—субъединиц LTCC - а также окислительными модификациями и нитрозилированием, которые изменяют кинетику стробирования и инактивации каналов [19]. Ранняя HF , подвергающаяся непрерывной β-адренергической стимуляции, запускает опосредованное протеинкиназой А (PKA) фосфорилирование субъединиц LTCC α_1, а также их вспомогательных β-субъединиц [4,8]. Острое фосфорилирование протеинкиназой А усиливает ток Са2+ L-типа, но хронические гиперадренергические состояния вызывают неадаптивное фосфорилирование, приводящее к десенсибилизации. Клетки HFrEF демонстрируют гиперактивированную активность CaMKII, которая модулирует фосфорилирование субъединиц LTCC для регуляции активности [8,20]. Фосфорилирование CaMKII повышает вероятность открытия и продлевает время открытия LTCC [8]. Активация CaMKII (CaMKII-опосредованный механизм, посредством которого повторный приток кальция увеличивает вероятность открытия и амплитуду тока Са2+-каналов L-типа) каналов приводит к более медленной инактивации и снижению общего тока Са2+-каналов L-типа [8]. Эти изменения приводят к ранней аритмогенной последующей деполяризации в поврежденных клетках [6]. Миоциты HFrEF обычно демонстрируют HF иженное функциональное количество LTCC, но более сильное фосфорилирование протеинкиназой A и CaMKII наряду с неправильным стробированием каналов [8].
Поразительной особенностью HFrEF является дисфункция связи EC (или “расцепление”) из-за структурного ремоделирования. В здоровых миоцитах LTCC на поперечных канальцах (T-канальцах) тесно взаимодействуют с рианодиновыми рецепторами (RyR2) на соединительных SR, обеспечивая синхронизированное высвобождение Ca 2+. HFrEF связан с разрушением и потерей Т-канальцев [4]. Исследования показали, что поврежденные желудочковые клетки образуют разреженную, дезорганизованную сеть Т-канальцев, в результате чего образуется “осиротевший” RyR2, который физически удален от мест запуска LTCC (рис. 1) [18]. Это приводит к задержке, диссинхронному высвобождению Ca 2+ и снижению вероятности успешного открытия LTCC, активирующего RyR2 [4]. Например, при постинфарктной HFrEF вероятность возникновения Ca 2+-искры и синхронность заметно снижаются, а задержка высвобождения Ca 2+ увеличивается, что согласуется с дефектной связью LTCC–RyR [4]. Ремоделирование Т-канальцев при HFrEF было причинно связано с этими изменениями — повышенное напряжение стенки и опосредованная растяжением сигнализация в дилатированных сердцах приводят к потере Т-канальцев и дисфункции кавеолина-3, тем самым нарушая передачу сигналов в микродоменах LTCC [5,17,18]. В результате кардиомиоциты HFrEF демонстрируют пониженное усиление связи с EC (меньше SR Ca 2+ высвобождается на единицу тока LTCC) и сниженную сократительную эффективность [4]. Этот механизм разобщения является ключевым фактором систолической дисфункции при HF [17].
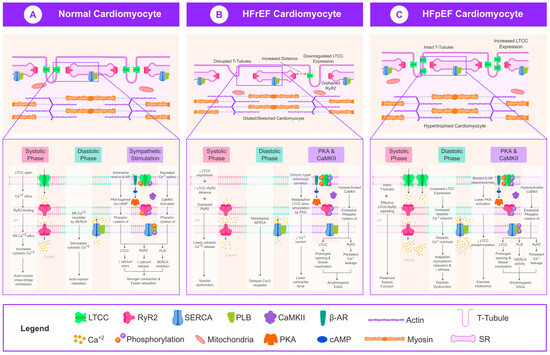 Figure 1. Schematic representation of L-type calcium channel (LTCC) behavior in normal, HFrEF, and HFpEF cardiomyocytes. (A) Normal cardiomyocyte showing organized T-tubules and tightly coupled LTCC–RyR2 interactions for efficient systolic Ca2+ release and diastolic reuptake. (B) HFrEF cardiomyocyte with disrupted T-tubules, increased LTCC–RyR2 distance, downregulated LTCC expression, maladaptive phosphorylation, and delayed Ca2+ reuptake—leading to systolic dysfunction and arrhythmogenesis. (C) HFpEF cardiomyocyte with preserved or increased LTCC expression, intact T-tubules, elevated diastolic Ca2+, and blunted Я-adrenergic responsiveness—leading to preserved EF but impaired relaxation. The figure illustrates phase-specific Ca2+ flux, LTCC gating, kinase signaling, and EC coupling efficiency across phenotypes. Abbreviations: LTCC, L-type calcium channel; RyR2, ryanodine receptor 2; SERCA, sarcoplasmic reticulum Ca2+-ATPase; PLB, phospholamban; CaMKII, calcium/calmodulin-dependent protein kinase II; Я-AR, beta-adrenergic receptor; PKA, protein kinase A; cAMP, cyclic AMP; HFrEF, heart failure with reduced ejection fraction; HFpEF, heart failure with preserved ejection fraction; EADs, early afterdepolarizations. Figure 1. Schematic representation of L-type calcium channel (LTCC) behavior in normal, HFrEF, and HFpEF cardiomyocytes. (A) Normal cardiomyocyte showing organized T-tubules and tightly coupled LTCC–RyR2 interactions for efficient systolic Ca2+ release and diastolic reuptake. (B) HFrEF cardiomyocyte with disrupted T-tubules, increased LTCC–RyR2 distance, downregulated LTCC expression, maladaptive phosphorylation, and delayed Ca2+ reuptake—leading to systolic dysfunction and arrhythmogenesis. (C) HFpEF cardiomyocyte with preserved or increased LTCC expression, intact T-tubules, elevated diastolic Ca2+, and blunted Я-adrenergic responsiveness—leading to preserved EF but impaired relaxation. The figure illustrates phase-specific Ca2+ flux, LTCC gating, kinase signaling, and EC coupling efficiency across phenotypes. Abbreviations: LTCC, L-type calcium channel; RyR2, ryanodine receptor 2; SERCA, sarcoplasmic reticulum Ca2+-ATPase; PLB, phospholamban; CaMKII, calcium/calmodulin-dependent protein kinase II; Я-AR, beta-adrenergic receptor; PKA, protein kinase A; cAMP, cyclic AMP; HFrEF, heart failure with reduced ejection fraction; HFpEF, heart failure with preserved ejection fraction; EADs, early afterdepolarizations.
Таким образом, HFrEF характеризуется снижением экспрессии/функции LTCC и структурным расцеплением LTCC с RyR2 (рисунок 1). Хроническая нейрогуморальная активация приводит к изменению фосфорилирования LTCC (например, с помощью CaMKII и протеинкиназы А), которое первоначально может быть компенсаторным, но в конечном итоге становится неадаптивным. Эти молекулярные и структурные изменения приводят к подавлению систолического высвобождения Ca2+ и сократительной способности [3,4], а также создают нестабильную среду Ca2+, которая предрасполагает к аритмиям (из-за LTCC и RyR2-опосредованной триггерной активности) [8]. Терапия HFrEF, такая как β-адреноблокаторы и механическая разгрузка, вероятно, частично приносит пользу сердцу, обращая вспять некоторые из этих изменений за счет повышения плотности LTCC, восстановления структуры Т-канальцев и уменьшения дисфункции каналов, вызванной гиперфосфорилированием [5,16].
3.2. LTCC Dysregulation in HFpEF
Регуляция LTCC при HFpEF категорически отличается. В отличие от HFrEF, при HFpEF систолическая функция сохраняется сравнительно хорошо (фракция выброса (EF) равняется или более 50%). В результате этого экспрессия LTCC и приток Са2+ также сохраняются сравнительно хорошо. Исследование, проведенное Kilfoil и соавт. показывает, что плотность LTCC и ток Са2+ в клетках L-типа сохраняются или даже слегка увеличиваются в моделях HFpEF по сравнению с нормой [4]. В модели крыс с гипертензивной HFpEF исходные миоциты демонстрировали большую амплитуду тока Ca2+ L-типа и амплитуду переходного процесса Ca2+, чем в контрольной группе. Вероятность искрового высвобождения Ca2+ при активации LTCC была увеличена, а задержка между открытием LTCC и высвобождением Ca2+ была уменьшена в клетках HFpEF, что указывает на более эффективную передачу сигналов LTCC-RyR2 по сравнению с HFrEF. Эти уникальные приспособления позволяют кардиомиоцитам HFpEF сохранять систолический выброс Ca2+ в условиях диастолической дисфункции (рис. 1) [1].
В соответствии с этим, исследование с использованием оптической визуализации при HFpEF у человека показало, что опосредованное LTCC высвобождение Ca2+ также хорошо сохраняется. В миоцитах пациентов с HFpEF, страдающих недостаточностью, отсутствовала выраженная десинхрония высвобождения Ca2+, характерная для HFrEF, и кратковременные выбросы Ca2+ в миоцитах, страдающих недостаточностью, были относительно скоординированными [5]. Данные в этом случае свидетельствуют о том, что миоциты HFpEF демонстрируют нормальное количество LTCC и функционируют, что позволяет общему показателю EF быть нормальным или близким к нормальному.
Структура поперечных канальцев остается почти нормальной при HFpEF по сравнению с обширным повреждением Т-образных канальцев, наблюдаемым при HFrEF. Как у людей, так и у животных с HFpEF плотность Т-канальцев в их уплотненных гипертрофированных сердцах была либо неизменена, либо увеличена, как показали гистологические исследования и визуализационные исследования [5]. Frisk и др. сообщалось, что кардиомиоциты человека с HFpEF, которые испытывали гипертрофические изменения без развития выраженной дилатации, сохраняли высокую плотность Т-канальцев и регулярную организацию, тем самым поддерживая эффективное взаимодействие LTCC–RyR [5]. Структурная целостность позволяет высвобождению Ca2+ оставаться синхронизированным с триггерными событиями LTCC в HFpEF. Обычная архитектура Т-образных трубочек в моделях HFpEF крыс обеспечивает почти нормальную искровую синхронизацию и время высвобождения Ca2+ [4]. HFpEF не проявляет признаков серьезного нарушения связи с EC, характерного для HFrEF, вместо этого клетки HFpEF обычно демонстрируют эффективную связь с EC на начальном этапе [4]. Повышенная вероятность срабатывания искры, наряду с более высокой синхронностью, наблюдалась при сверхнормативной точности сопряжения в одном исследовании, которое предложило это в качестве компенсаторного механизма для противодействия повышению жесткости стенки желудочка при HF [4]. Такая адаптация позволяет сохранить ударный объем при нарушенной релаксации [4].
В здоровых клетках сердечной мышцы поперечные канальцы или Т-канальцы образуют хорошо организованную сеть, которая гарантирует, что кальциевые каналы L-типа (LTCC) идеально согласованы с рианодиновыми рецепторами (RyR2) в саркоплазматическом ретикулуме. Эта тесная структурная взаимосвязь необходима для скоординированного высвобождения кальция, позволяющего сердцу эффективно сокращаться [12].
Напротив, сердечная недостаточность со сниженной фракцией выброса (HF ) нарушает эту структуру. По мере расширения сердца и истончения его стенок механическая нагрузка возрастает. Со временем это вызывает структурную перестройку, которая повреждает систему Т-образных канальцев [5,12]. На молекулярном уровне происходит подавление регуляции работы белков, ответственных за поддержание целостности Т-канальцев, таких как junctophilin-2 (JPH2) и amphiphysin-2 (BIN1). Кавеолин-3, еще один важный структурный белок, также проявляет признаки дисфункции при сердечной недостаточности [12]. В дополнение к этому цитоскелет становится нестабильным, а изменения в сети микротрубочек влияют на то, как LTCC доставляются к клеточной мембране и закрепляются в ней [12]. В результате образуется фрагментированная и неупорядоченная сеть Т-образных канальцев, часто пронизанная фиброзной тканью. Это приводит к разделению LTCC и RyR2s, состоянию, называемому “потерянным” RyR2s, которое нарушает кальциевую передачу сигналов и ослабляет силу сокращения [5,12].
Сердечная недостаточность с сохраненной фракцией выброса (HFpEF), однако, представляет собой другую историю. Вместо расширения камер, наблюдаемого при HFrEF, HFpEF обычно сопровождается концентрическим утолщением стенки сердца. Это ремоделирование помогает нормализовать напряжение в стенке и, по-видимому, защищает систему Т-образных канальцев от повреждений, наблюдаемых при HFrEF [5,12]. Фактически, данные, полученные как на экспериментальных моделях, так и при биопсии пациентов, показывают, что сеть Т-канальцев при HFpEF часто сохраняется, а в некоторых случаях даже слегка расширяется. Адаптивные реакции, такие как увеличение образования канальцев или их небольшое расширение, могут помочь сохранить их целостность [5,12]. Благодаря этому кальциевые каналы и рецепторы остаются тесно связанными, поддерживая нормальное высвобождение кальция и сохраняя систолическую функцию.
Таким образом, в то время как HFrEF характеризуется структурными нарушениями и пропусками кальциевых сигналов, HFpEF в значительной степени сохраняет свою архитектурную структуру, позволяя сердцу сохранять свою насосную способность. Эти различия подчеркивают тот факт, что HFrEF и HFpEF - это не просто точки в одном и том же спектре, а различные состояния с различными лежащими в их основе механизмами [5].
Несмотря на то, что функция LTCC сохраняется, HFpEF можно рассматривать как заболевание с неадекватной передачей сигналов и диастолическим контролем Са2+ [2]. Следует отметить, что при HFpEF наблюдается замедленное расслабление, а также повышенное диастолическое напряжение [21]. На уровне отдельных клеток диастолическое содержание Ca2+ в кардиомиоцитах HFpEF повышено [4].Kilfoil и соавт. отметили, что миоциты крыс с HFpEF имели повышенный уровень внутриклеточного кальция, что, в свою очередь, увеличивало жесткость миоцитов и напряжение в состоянии покоя [4]. Повышенные диастолические уровни Ca2+ не были вызваны серьезным нарушением обратного захвата SR Ca2+; в этом исследовании уровни кальциевой атфазы 2a саркоплазматического/эндоплазматического ретикулума (SERCA2a) и фосфорилирования фосфоламбана (PLB) не были изменены при HFpEF [4]. Вместо этого авторы предполагают, что увеличение притока Ca2+ через LTCC и утечки Ca2+ из каналов RyR2 может увеличить диастолическое значение Ca2+ при HFpEF [4]. То есть выброс Ca2+ во время систолы является нормальным, но во время диастолы наблюдается умеренная перегрузка Ca2+ в результате небольшого увеличения тока LTCC и утечки SR Ca2+ [4]. Этот путь регулируется Ca2+/кальмодулин-зависимой протеинкиназой II (CaMKII), которая фосфорилирует LTCCs и RyR2 для увеличения притока Ca2+ и диастолической утечки Ca2+ [8]. Повышенный приток Ca2+ в SR увеличивает нагрузку SR на Ca2+, что вместе с сенсибилизацией к RyR2 приводит к неадекватному высвобождению Ca2+ во время диастолы [8]. Утечка, опосредованная RyR2, вероятно, вызвана повышенным фосфорилированием в сайте Ser2808, посттрансляционной модификацией, которая увеличивает вероятность открытия RyR2 без существенного истощения запасов SR Ca2+ [4]. Диастолическая утечка Ca2+, опосредованная RyR2, приводит к повышению уровня Ca2+ в цитозоле в состоянии покоя и активирует поступающие в обменник Na+/Ca2+ потоки, которые нарушают гомеостаз кальция и стабильность мембран. Повышенная диастолическая активность обмена Ca2+ и Na+/Ca2+ приводит к отсроченным постдеполяризациям, которые представляют собой низкоамплитудные деполяризации, которые могут вызывать аритмии, когда сердце находится в уязвимом состоянии [8]. Это открытие согласуется с другими исследованиями, которые показали, что HFpEF (особенно при наличии метаболических заболеваний, таких как диабет) связан со снижением выброса Ca2+ и повышением диастолического уровня Ca2+ в миоцитах [5]. Нарушение диастолической регуляции Ca2+ при HFpEF приводит к нарушению расслабления и повышению жесткости миокарда; связанный с тропонином Ca2+ увеличивает напряжение в состоянии покоя, а снижение фосфорилирования титина (из-за снижения активности протеинкиназы G (PKG)) увеличивает жесткость кардиомиоцитов [4]. Следовательно, HF можно рассматривать как нарушение нормальной функции LTCC во время систолы и нарушение диастолической регуляции Ca2+
Другим аспектом регуляции LTCC при HFpEF является притупление β-адренергической реакции. Клинически пациенты с HFpEF имеют низкую физическую работоспособность и хронотропный/инотропный резерв. Исследования показывают, что на клеточном уровне миоциты с HFpEF не способны эффективно усиливать циркуляцию Ca2+ при β-адренергической стимуляции [4]. В вышеупомянутой модели на крысах isoproterenol (β-агонист) вызывал значительно меньшее увеличение амплитуды переходного процесса Ca2+ и обратного захвата SR Ca2+ клетками HFpEF по сравнению с контролем [4]. Это свидетельствует о десенсибилизации β-адренергической сигнализации при HFpEF. Возможные механизмы включают подавление или отсоединение β1-адренорецепторов, усиление регуляции ингибирующих β3-рецепторов или нарушение выработки цАМФ в сердце при HFpEF [4,5]. Конечным результатом является то, что фосфорилирование LTCC протеинкиназой А при стрессе подавляется, что ограничивает усиливающий эффект на ток Ca2+ L-типа и обратный захват Ca2+. По сути, сердца при HFpEF, вероятно, работают в режиме, близком к максимальному использованию Са2+ в исходном состоянии (для сохранения EF), оставляя небольшой запас, когда потребность в адренергии возрастает [4]. Этот “предельный эффект” (“ceiling effect” ) способствует непереносимости физических нагрузок при HFpEF. Это также частично объясняет, почему методы лечения, эффективные при HFrEF (которые уменьшают чрезмерную β-стимуляцию), не показали пользы при HFpEF, поскольку HFpEF уже подавляет β-адренергическую сигнализацию [4].
Сохраненная экспрессия LTCC и исходная функция позволяют HFpEF демонстрировать нормальный систолический выброс Ca 2+; это противоположно HFrEF, который демонстрирует дефицит LTCC [4,5]. Клетки HFpEF также плохо адаптируются к обработке Ca 2+ и передаче сигналов в диастолу, однако у них повышен уровень Ca 2+ в состоянии покоя и снижен β-адренергический резерв [4]. Эти молекулярные различия позволяют понять, что HFpEF - это не просто "легкая форма HFrEF", а скорее отдельное заболевание, которое, как правило, вызвано системным воспалением, окислительным стрессом и гипертрофическим ремоделированием, а не серьезным истощением клеточного Ca 2+, наблюдаемым при HFrEF [5,22]. Краткое параллельное сравнение этих фенотипических особенностей представлено в таблице 1.
Таблица 1. Краткое изложение ключевых молекулярных, структурных и функциональных различий в опосредованной LTCC обработке Ca 2+ между кардиомиоцитами HFrEF и HFpEF. 3.3. Role of CaMKII in LTCC Regulation
Ca2+/кальмодулин-зависимая протеинкиназа II (CaMKII) стала важным регулятором сердечных ионных каналов наряду с Ca2+. В сердечных миоцитах CaMKII интегрирует сигналы Ca2+ и окислительного стресса [23]. Наряду с этим, она может изменять функцию LTCC посредством фосфорилирования и других механизмов [22]. Как HFrEF, так и HFpEF демонстрируют повышенную активацию CaMKII, однако последствия для регуляции LTCC могут отличаться.
В физиологических условиях CaMKII отвечает за калибровку активности LTCC посредством процесса, известного как Са2+-зависимое стимулирование [1,2]. Повторяющийся приток Ca2+ приводит к постепенному увеличению вероятности открытия LTCC и амплитуды тока, частично опосредованному фосфорилированием CaMKII субъединиц LTCC [8,19]. Молекулярные мишени включают участки на β-субъединице LTCC (например, Thr^498 на β_2a) и, возможно, субъединицу α_1C, которая при фосфорилировании CaMKII стабилизирует канал в режиме (gating) стробирования с высокой активностью [8]. Фосфорилирование, катализируемое CaMKII, способствует стробированию (gating) LTCC во 2-м режиме (дисфункциональное состояние каналов, характеризующееся длительным открытием и высокой вероятностью открытия, приводящее к чрезмерному притоку Ca2+), которое характеризуется более длительным открытием и более высокой вероятностью открытия [8]. Это приводит к увеличению притока Ca2+ для данной деполяризации, но также замедляет инактивацию тока Ca2+ L-типа [8]. В условиях заболевания чрезмерная активность CaMKII может ненадлежащим образом перевести LTCC в этот облегченный режим. На самом деле, гиперактивное gating LTCC из-за CaMKII наблюдалось при сердечной недостаточности [20]. Mork и др. сообщали, что одиночные LTCC из поврежденных миоцитов человека показали более высокую вероятность открытия и доступность (в соответствии с CaMKII-опосредованным упрощением), несмотря на уменьшение общего числа каналов [11,19]. Это управляемое CaMKII усиление раскрытия LTCC способствует аритмогенной деполяризации: стробирование (gating) в режиме 2 и поздние токи Ca2+ могут ускорять ранние постдеполяризации и вызывать эктопические биения [8]. Более того, увеличивая приток Ca2+, CaMKII может привести к перегрузке SR Ca2+ и последующей диастолической утечке Ca2+ через RyR2, способствуя отсроченной постдеполяризации [8].
Было обнаружено, что активность CaMKII повышается при сердечной недостаточности (HF ). Как у пациентов с HFpEF, так и у пациентов с HFrEF наблюдается повышенный уровень изоформ CaMKII? и повышенный уровень аутофосфорилирования [22]. Пациенты с HFrEF испытывают хроническую адренергическую стимуляцию и нарушают гомеостаз кальция, что приводит к избыточной экспрессии CaMKII, создавая цикл отрицательной обратной связи, который нарушает обработку кальция [8,22]. CaMKII поддерживает конститутивную активность посредством аутофосфорилирования по Thr^ 287 или окислительной модификации Met^ 281/282 в пределах своей регуляторной области [8,22]. У пациентов с HF с сопутствующими заболеваниями, такими как сахарный диабет и ожирение, отмечаются повышенные уровни активных форм кислорода (АФК), которые определяют окислительный стресс как характерный признак HF . Повышенные уровни АФК вызывают окисление CaMKII при Met^ 281/282, что приводит к длительной активности фермента, даже когда уровень кальция нормализуется [8,22]. Патологическое ремоделирование было связано с окисленной формой CaMKII, поскольку у мышей, экспрессирующих устойчивый к окислению CaMKII (Met281/282Val), развивается защита от индуцированной ангиотензином II гипертрофии и сердечной недостаточности [22]. CaMKII функционирует как важное связующее звено между ?-адренергическими сигналами и окислительно-восстановительным стрессом для контроля активности LTCC [24]. Системное воспаление и активные формы кислорода могут активировать CaMKII у пациентов с HFpEF, поскольку β-адренергическая стимуляция менее выражена, чем при HFrEF [22]. Полученные данные свидетельствуют о том, что CaMKII является ключевым интегратором между нейрогормональными (β-адренергическими) и окислительно-восстановительными механизмами стресса, которые регулируют активность LTCC.
Последующее воздействие гиперактивности CaMKII включает дисфункцию LTCC и неправильное обращение с Ca2+ [24]. При HFrEF сверхактивная CaMKII способствует снижению доступности Ca2+-каналов (за счет усиленной интернализации или изменения оборота каналов), но также, как это ни парадоксально, увеличивает поздние токи Ca2+ и риск аритмии из-за режима gating 2 [8,20]. CaMKII также фосфорилирует RyR2, усиливая утечку SR Ca2+ в HF, и фосфорилирует фосфоламбан (PLB) при Thr^17, что может изменять функцию SERCA [22]. При HFpEF CaMKII может играть роль в снижении β-адренергического резерва: хроническая активация CaMKII может индуцировать десенсибилизацию передачи сигналов β-рецепторами и способствовать изменению активности фосфодиэстеразы, ограничивая эффекты цАМФ/протеинкиназы А на LTCC [22,23]. Также имеются доказательства того, что CaMKIIδ перемещается в ядро и изменяет транскрипцию при HF, поддерживая программу экспрессии и гипертрофии генов плода, которая может косвенно модулировать экспрессию субъединицы LTCC или структуру Т-канальцев [22].
При недостаточности сердца человека экспрессия CaMKIIδ повышается, особенно варианта сплайсинга CaMKIIδ_B, который содержит сигнал ядерной локализации, который направляет его к ядру кардиомиоцита [25]. После транслокации ядерная CaMKIIδ_B запускает неадаптивные программы транскрипции, связанные с гипертрофией сердца и прогрессированием сердечной недостаточности. Ключевой механизм включает фосфорилирование гистондеацетилаз класса IIa (HDAC4 и HDAC5), которые впоследствии экспортируются из ядра. Это ослабляет их ингибирование фактора-энхансера миоцитов 2 (MEF2), фактора транскрипции, который способствует экспрессии гипертрофических генов [25].
Этот чувствительный к кальцию сигнальный каскад приводит к реактивации профиля экспрессии генов плода, характеризующегося повышением уровня предсердного натрийуретического фактора (Anf), натрийуретического пептида типа В (Bnp), тяжелой цепи β-миозина и скелетного α-актина [25]. В дополнение к модуляции гипертрофических генных программ, устойчивая активность CaMKII влияет на белки, переносящие кальций. Длительная активация была связана со снижением регуляции SERCA2a и фосфоламбана (PLB), а также с повышенной экспрессией изменений в обменнике Na+/Ca2+ (NCX1), которые способствуют внутриклеточной перегрузке кальцием и диастолической дисфункции [26].
В совокупности эти результаты показывают, что CaMKIIδ _B не только регулирует функцию ионных каналов, но и управляет транскрипционным перепрограммированием при сердечной недостаточности, связывая электрическую активность, сигналы стресса и долгосрочные изменения экспрессии генов.
Очевидно, что CaMKII является главным “злодеем” в патофизиологии HF [8,23]. Широкий спектр его мишеней — LTCC, RyR2, PLB и т.д. — означает, что гиперактивность CaMKII может одновременно ослаблять сократительную способность и увеличивать аритмогенный риск [8,24]. Например, было показано, что CaMKII-зависимое фосфорилирование LTCC и фосфорилирование RyR2 синергически способствуют ранней постдеполяризации в моделях HF [20]. Признание центральной роли CaMKII сделало его привлекательной терапевтической мишенью (обсуждается ниже). Текущие исследования также направлены на изучение того, как конкретно прервать взаимодействие CaMKII с комплексами LTCC — например, предотвратить доступ CaMKII к субъединице LTCC β_2 или ингибировать ее окисление — в качестве средства, с помощью которого можно нормализовать поведение LTCC при сердечной недостаточности [8,20].
CaMKII действует как критический модификатор функции LTCC при HF. Он становится устойчиво активным как при HFrEF, так и при HFpEF, что приводит к гиперфосфорилированию LTCC. Это приводит к увеличению вероятности открытия LTCC и замедлению инактивации (facilitation) [8], что может усугубить нарушение регуляции Ca2+, способствуя возникновению аритмий (вызывая аберрантную деполяризацию) и сократительной дисфункции (истощая SR Ca2+ или нарушая расслабление) [4,8,15]. Таким образом, нацеливание на CaMKII и его последующее воздействие на LTCC представляет собой логичную стратегию улучшения сердечной функции при HF .
3.4. Emerging Therapeutic Targets
Учитывая ключевую роль дисфункции LTCC и CaMKII в патогенезе HF , несколько новых терапевтических стратегий направлены на нормализацию регуляции LTCC или ее последующих последствий. Традиционные методы лечения HF (β-адреноблокаторы, ингибиторы ренин–ангиотензиновой системы (RAS)) обеспечивают косвенную пользу, снижая хронический адренергический и гемодинамический стресс, но более новые подходы направлены на более прямое воздействие на механизмы регуляции Ca2+ в сердце.
3.4.1. CaMKII Inhibition
Поскольку CaMKII является молекулярным компонентом, усугубляющим как систолический, так и диастолический дефицит Ca2+, он стал главной мишенью для разработки лекарств [8,22]. Доклинические исследования с использованием генетического ингибирования CaMKII или пептидных ингибиторов (таких как autocamtide inhibitory peptide (AIP) и пептид-ингибитор CaMKII (CaMKIIN)) показали улучшение сердечной функции и уменьшение аритмий на моделях HF [22]. До недавнего времени CaMKII считался "нелекарственным" из-за его повсеместного применения, но появились новые низкомолекулярные ингибиторы, такие как autocamtide inhibitory peptide (AIP), CaMKIIN, AS105 и hesperadin, которые демонстрируют избирательную эффективность на моделях сердца [27,28]. В экспериментальных моделях HFpEF, характеризующихся высоким окислительным стрессом, ингибирование окисления CaMKII, достигаемое за счет сверхэкспрессии метионинсульфоксид-редуктазы или за счет мутации Met281/282 в CaMKII, это способствовало сохранению диастолической функции. Это подчеркивает терапевтический потенциал воздействия на окислительно-восстановительную активацию CaMKII [22]. Ингибиторы CaMKII, такие как аутокамтидный ингибирующий пептид (AIP), CaMKIIN, AS105 и гесперидин, могут обеспечить двойную пользу при сердечной недостаточности, повышая сократительную способность за счет стабилизации циркуляции Ca2+ и снижая риск аритмий.Это было бы особенно полезно для пациентов с HF , у которых в настоящее время отсутствуют эффективные методы лечения, за счет прямого воздействия на нарушение регуляции Ca2+, вызванное воспалением и активацией CaMKII — механизмов, которые не поддаются эффективному лечению традиционными методами лечения сердечной недостаточности или неспецифическими противовоспалительными средствами [29,30].
3.4.2. LTCC Modulation
Прямая модуляция активности LTCC представляет собой еще одну стратегию. Применение дигидропиридинов, таких как amlodipine представляет собой традиционные блокаторы LTCC, которые служат полезными антигипертензивными и антиангинальными препаратами, но оказывают неблагоприятное влияние на систолическую функцию при HF , дополнительно снижая поступление Са2+. Возрос интерес к разработке более точных модуляторов LTCC. Один из методов заключается в воздействии на вспомогательные белки, которые регулируют транспортировку LTCC и их формирование. Например, малая Rad GTPase обычно ограничивает LTCC, и его уровни снижаются при HFrEF, в результате чего вероятность открытия остальных каналов увеличивается [20]. Стратегии, направленные на восстановление функции Rad, могут способствовать модуляции активности LTCC, не влияя на экспрессию каналов. Другой потенциальной стратегией является сосредоточение внимания на определенных подтипах LTCC, например, Cav1.3, который представляет собой незначительную изоформу канала L-типа, которая повторно экспрессируется в желудочках с недостаточностью и может вызывать аритмогенные медленные Ca2+-токи [11]. Селективное ингибирование Cav1.3, хотя все еще находится на доклинической стадии, может предложить будущую стратегию профилактики аритмий при HF без ущерба для Cav1.2-зависимой сократительной способности [31]. Кроме того, результаты изучения природных токсинов, таких как maitotoxin и atrotoxin, которые активируют кальциевые каналы, являются ценными моделями для разработки агонистов LTCC. Хотя эти токсины не являются терапевтически жизнеспособными из-за их высокой токсичности, они помогают идентифицировать потенциальные сайты проникновения и конформационные изменения, которые могли бы вдохновить на разработку будущих лекарств-агонистов [32-36].
3.4.3. RyR2 Stabilizers and Ca2+ Cycling Enhancers
Хотя воздействие на канал высвобождения SRCa2+ является одним из этапов после LTCC, оно напрямую влияет на связь между триггером LTCC и выходом SR Ca2+. Соединения, известные как Rycals (например, ARM210), были разработаны для стабилизации RyR2 в закрытом состоянии, уменьшая диастолическую утечку Ca2+ [2,3]. Устраняя “негерметичный клапан” SR, эти препараты могут улучшить содержание Са2+ в SR и его сократительную способность, одновременно снижая аритмогенные выбросы Са2+. Стабилизатор RyR2 (например, elamipretide) может дополнять нормализацию LTCC — LTCC обеспечивает запуск, а более здоровый SR обеспечивает более сильную реакцию Ca2+ без утечки. Некоторые препараты Rycals прошли клинические испытания для лечения HF или катехоламинергического полиморфного VT [3]. Аналогичным образом, повышение активности SERCA2a с помощью генотерапии (Ad-SERCA2a, протестировано в исследовании CUPID) было попыткой улучшить обратный захват Ca2+ и сократительный резерв при HFrEF [3]. Хотя первоначальные результаты генотерапии SERCA у людей были скромными, в настоящее время проводятся усовершенствования (например, целенаправленная доставка или комбинированная модуляция PLB). Эти подходы, хотя и не воздействуют непосредственно на LTCC, влияют на ту же ось связи EC и, таким образом, имеют отношение к общему гомеостазу Са2+ при HF .
4. Future Directions
Лечение HFrEF могло бы выйти за рамки существующей стандартной практики, разработав препараты, которые активируют ток LTCC либо только во время систолы, либо без диастолической активации, чтобы увеличить сократительную способность при меньших утечках в состоянии покоя. Генотерапия или подходы на основе РНК, которые усиливают экспрессию BIN1 или junctophilin-2, могли бы создать более эффективную связь LTCC-RyR в поврежденных сердцах. Исследования должны быть направлены на выявление факторов, способствующих дисфункции LTCC, поскольку ингибиторы CaMKII потребуют клинических испытаний в качестве первого шага к созданию новых методов лечения HF , основанных на молекулярных субстратах аритмий. LTCC-субъединица Cav1.3, наряду с другими LTCC-субъединицами, присутствующими при HF, демонстрирует потенциал для будущих исследований по разработке специализированных регуляторов, которые могли бы точно регулировать сократительную способность и возбудимость сердца. Исследование HFpEF требует срочного изучения того, как метаболические и воспалительные факторы влияют на механизмы регуляции кальция кардиомиоцитами. Модели HFpEF на крупных животных вместе с кардиомиоцитами, полученными из iPCS у пациентов с HFpEF, позволяют ученым изучить, как сопутствующие заболевания влияют на функцию LTCC и механизмы циркуляции Ca2+. Медицинское сообщество проявляет растущий интерес к разработке методов лечения специфических профилей HFpEF [21]. Антицитокиновая или антиоксидантная терапия могла бы лечить HF, опосредованную воспалением, путем нормализации функции CaMKII и LTCC, но пациентам с HF , основной причиной которых является артериальная гипертензия, были бы полезны фенотипически специфичные модуляторы передачи Са2+ или ингибиторы киназы легких цепей миозина (MLCK) для усиления релаксации [21]. Сочетание механической и молекулярной терапии открывает новое направление, поскольку физические нагрузки и инновационные наполнители, используемые в сочетании с фармакологическими препаратами, такими как ингибиторы натрий-глюкозного котранспортера-2 (SGLT2) или агонисты гуанилатциклазы, обеспечивают синергетический эффект для регуляции Ca2+ и диастолической функции. Подходы Omics и системной биологии помогут идентифицировать новые регуляторные элементы функции LTCC с помощью микроРНК и каркасных белков, которые изменяют экспрессию во время HF. Лечение этих вышестоящих регуляторов дает возможность устранить первоначальную дисфункцию, связанную с обработкой Ca2+. Недавние научные результаты показывают, что микроРНК подавляют субъединицы LTCC у пациентов с HF посредством механизмов репрессии, которые могут быть использованы для изменения экспрессии LTCC при HFrEF и уменьшения избыточного притока Ca2+ при HFpEF.
Применение терапевтических методов лечения, основанных на молекулярных исследованиях, требует точной корректировки, чтобы избежать нарушения баланса Са2+, необходимого для поддержания сердечного ритма. В будущем для лечения HF потребуются точные медицинские подходы, позволяющие определить, у каких пациентов наблюдается дефицит Са2+ или проблемы с контролем Са2+, чтобы врачи могли назначить соответствующее лечение. Медицинские работники могут разрабатывать индивидуальные методы лечения сердечно-сосудистых заболеваний, изучая HFrEF и HFpEF на молекулярном уровне, включая регуляцию LTCC и ее последующие эффекты. Эффективность лечения HFrEF по сравнению с HFpEF будет установлена в ходе текущих и предстоящих клинических испытаний, в ходе которых будут изучены микродомены CaMKII и LTCC, а также управление уровнем диастолического Ca2+. Исследования регуляции LTCC при HF показывают потенциал для разработки более эффективных методов лечения различных клеточных аномалий, обнаруженных при HFrEF и HFpEF.
В соответствии с новыми исследованиями, будущие терапевтические стратегии при HF все больше фокусируются на точном воздействии на нарушенную регуляцию Ca2+ и активность CaMKII. Например, в настоящее время разрабатываются высокоселективные ингибиторы CaMKII: первые в своем классе молекулы Cardurion (например, CRD-2015, CRD-2959) продемонстрировали мощное ингибирование CaMKII-мишеней и улучшенную выживаемость в доклинических моделях HF [37], а ведущий препарат уже прошел испытания в фазе 1. (с планами расширения на показания к HF ) [37]. Также предпринимаются усилия по вмешательствам, специфичным для микродоменов, которые стабилизируют локальные сигнальные комплексы Ca2+; в redoxчастности, доставка гена организатора Т-канальцев cBIN1 восстанавливала dyadic микродомены и обращала вспять фенотипы сердечной недостаточности на животных моделях [38]. Учитывая патологическую redox-активацию CaMKII при HF , изучаются новые redox-чувствительные подходы, включая точное редактирование генов для устранения чувствительных к окислению метионинов CaMKII [39] или даже его участка аутофосфорилирования [40], что в недавних доклинических исследованиях дало кардиопротекторные эффекты.
Генная терапия нового поколения, направленная на обработку кальцием, набирает обороты: например, недавно у пациентов с HFpEF (впервые у человека) был протестирован генный перенос гена SERCA2a, опосредованный AAV1 (аденоассоциированный вирус серотипа 1), который показал отсутствие серьезных проблем с безопасностью и раннее улучшение физической активности и сердечных биомаркеров [41]. Аналогичным образом, редактирование на основе CRISPR/Cas9 патогенных регуляторов кальция (например, CaMKIIδ) оказалось возможным на кардиомиоцитах и мышах, что позволяет предположить постоянный способ подавления неадаптивной передачи сигналов Ca2+ [39]. Между тем, в настоящее время активно исследуются методы терапии на основе РНК для точной настройки гомеостаза Ca2+ в миокарде, например, антисмысловое ингибирование микроРНК-132 (CDR132L) было безопасным в фазе 1 и продемонстрировало обнадеживающие улучшения сердечной функции у пациентов с HF [42]. Наконец, кардиомиоциты, полученные из iPSC, специфичные для конкретного пациента, стали платформой для тестирования лекарственных препаратов и персонализированного моделирования HF , что позволяет проводить высокопроизводительный скрининг методов лечения на кардиальных клетках, подобранных для пациентов [43]. Эти перспективные подходы, которые в настоящее время распространяются от лабораторных исследований к клиническим, открывают более перспективные направления лечения, нацеленные на CaMKII и на нарушение регуляции кальция при сердечной недостаточности.
5. Conclusions
Две различные парадигмы нарушения регуляции LTCC проявляются в виде сердечной недостаточности со сниженной и сохраненной E . Сочетание хронического стресса приводит к тому, что пациенты с HFrEF теряют плотность LTCC, в то время как их связь LTCC–RyR нарушается, что приводит к ослаблению связи EC и систолической недостаточности. Чрезмерная активация киназ PKA и CaMKII при HFrEF приводит к гиперфосфорилированию LTCCs и RyR2, что приводит к утечке Ca2+ и аритмиям. Сердце поддерживает свой систолический выброс за счет компенсации HFpEF, даже несмотря на то, что экспрессия LTCC и исходная функция остаются в основном неизменными. Миоциты пациентов с HFpEF испытывают дезадаптивную сигнализацию в виде повышенного диастолического уровня Са2+, ригидности миоцитов и снижения β-адренергической реакции, что ухудшает расслабление и резерв. Фермент CaMKII функционирует как общее связующее звено между патологическими модификациями LTCC, которые вызывают как дисфункцию LTCC при HFrEF, так и повышенный диастолический уровень Ca2+ при HFpEF. Различные механизмы заболевания объясняют, почему лечение HFrEF, снижающее симпатическую активность и обратное ремоделирование, не привело к лучшим результатам у пациентов с HFpEF.
|