Приблизительно 1 из 1000 детей имеет тяжелую или полную потерю слуха при рождении или в раннем детстве, что определяется как pre-lingual глухота (1). Более 50% pre-lingual глухоты в развитых странах связано с моногенными дефектами (2).Относительная пропорция генетических случаев увеличивается со временем улучшения условий здоровья общества, что ведет к снижению превалирования потери слуха от негенетических причин, таких как инфекция (2). Генетическая глухота подразделяется на синдромальные формы, в которых потеря слуха ассоциирует с др. разнообразными аномалиями. Синдромальные формы составляют 30% pre-lingual генетической глухоты и включают несколько сотен синдромов глухоты (3), с генетическими основами, обнаруженными примерно у 30 из них (4, 5).
При не-синдромальной генетической глухоте с pre-lingual началом, предоминирует аутосомно-рецессивное наследование (80%), над аутосомно-доминантными (20%), X-сцепленными (1%) и митохондриальными (менее 1%) формами (1). При post-lingual, не-синдромальной глухоте аутосомно-рецессивное наследование очень редко. Аутосомно-рецессивные формы обычно более тяжелые, чем др. и почти все ведут к дефектам улитки (нейросенсторная глухота). Не-синдромальная глухота генетически чрезвычайно гетерогенна, т.к. картировано 85 локусов и идентифицировано 39 ядерных генов (December 2005), включая 23 DFNB генов (Van Camp G, Smith RJH. Hereditary Hearing Loss. http://www.dnalab-www.uia.ac.be/dnalab/hhh). За последнее время опубликовано несколько обзоров (4-14), но новые гены обнаруживаются со 'скоростью звука', мы представим обновленный список локусов и генов и рассмотрим вклад в глухоту в разных популяциях. Недавно мы представили обзор по аутосомно-доминантной глухоте (15) поэтому сфокусируемся тут на аутосомно-рецессивной глухоте.
Локусы и гены не-синдромальной, аутосомно-рецессивной глухоты представлены в Tables 1 и 2. Всего 23 разных гена было идентифицировано и они описаны кратко ниже.
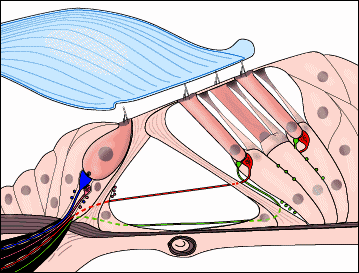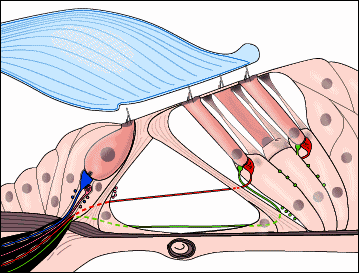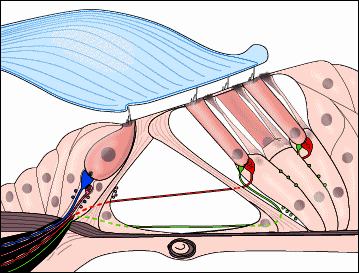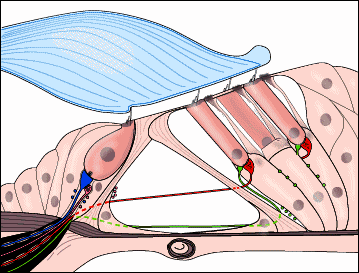
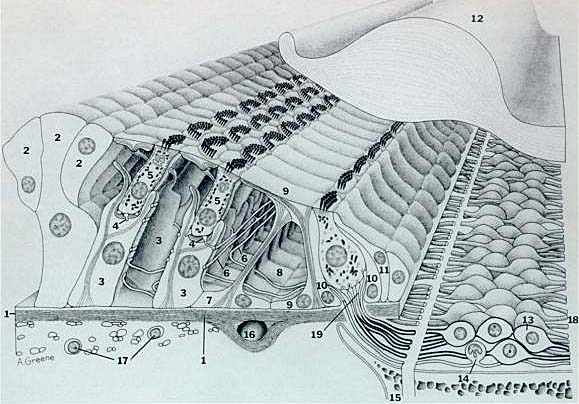 Разрез типичного базального витка улитки млекопитающих. Диаметр наружной волосковой клетки примерно 7µm. Пустые пространства в основаниях наружных волосковых клеток заняты эфферентными окончаниями, которые не видны на рис. 1 - базилярная мембрана, 2 - Hensen клетки, 3 - наружные фалангеальные клетки Deiters, 4 - нервные окончания, 5 - наружные волосковые клетки, 6 - наружные спиральные волокна, 7 - наружные pillar клетки, 8 - внутренний туннель, 9 - внутриенние pillar клетки, 10 - внутренние фалангиальные клетки, 11 - пограничные клетки, 12 - текториальная мембрана, 13 - клетка спирального ганглия типа I, 14 - клетка спирального ганглия типа II, 15 - костная спиральная ламина, 16 - спиральный кровеносный сосуд, 17 - веретенообразные клетки, 18 - аксоны клетки спирального ганглия (волокна слухового нерва), 19 - радиальные волокна.
Разрез типичного базального витка улитки млекопитающих. Диаметр наружной волосковой клетки примерно 7µm. Пустые пространства в основаниях наружных волосковых клеток заняты эфферентными окончаниями, которые не видны на рис. 1 - базилярная мембрана, 2 - Hensen клетки, 3 - наружные фалангеальные клетки Deiters, 4 - нервные окончания, 5 - наружные волосковые клетки, 6 - наружные спиральные волокна, 7 - наружные pillar клетки, 8 - внутренний туннель, 9 - внутриенние pillar клетки, 10 - внутренние фалангиальные клетки, 11 - пограничные клетки, 12 - текториальная мембрана, 13 - клетка спирального ганглия типа I, 14 - клетка спирального ганглия типа II, 15 - костная спиральная ламина, 16 - спиральный кровеносный сосуд, 17 - веретенообразные клетки, 18 - аксоны клетки спирального ганглия (волокна слухового нерва), 19 - радиальные волокна.
GJB2 (connexin 26) - DFNB1
Наиболее важный локус для не-синдромальной аутосомно-рецессивной глухоты (DFNB1) первоначально был найден на хромосоме 13q11 путем анализа сцепления в двух крупных близкородственных тунисских семьях с pre-lingual, выраженной глухотой (16). Последующие исследования сцеплений в New Zealand/Australian (17) и Italian/Spanish семьях с глухотой (18) показали. что этот локус является главным вкладчиком в pre-lingual глухоту. Мутации в гене GJB2, кодирующем белок щелевых соединений connexin 26, который был картирован в 13q11-q12 (19), затем были идентифицированы в трех родственных семьях в Пакистане с выраженной глухотой, генетически сцепленной с 13q11 (20). Ген GJB2 был первымt DFNB геном идентифицированным в 1997.
Ген GJB2 обладает единственным кодирующим экзоном, а белок принадлежит к большому семейству коннексинов, имеющих 4 трансмембранных домена, которые участвуют в межклеточных коммуникациях посредством щелевых соединений (21). 6 субъединиц коннексина вместе формируют гексамер (connexon) в плазматической мембране и каждый коннексон ассоциирует с др. коннексоном на соседней клетке, чтобы сформировать межклеточный канал; а множественные каналы, в свою очередь, образуют кластеры в специализированной области мембраны, чтобы сформировать щелевые соединения. Коннексоны могут быть гомомерными (состоящими из идентичных коннексиновых субъединиц) или гетеромерными ( состоящими из более чем одного вида конексинов) (21). Коннексины важны для рециклинга ионов калия в эндолимфе улитки посредством сети щелевых соединений, которые идут от эпителиальных поддерживающих клеток к fibrocytes спиральной лигаменты и к эпителиальным маргинальным клеткам stria vascularis (22, 23). Гомеостаз ионов является важным для нормального слуха, а мутации в некоторых генах, кодирующих коннексины, или ионные каналы, ведут к наследственной глухоте (22-24).
Мутации в гене GJB2 являются основной причиной pre-lingual, не-синдромной рецессивной глухоты, т.к. они ответственны за более чем 50% таких случаев во многих популяциях (24-30). Одна специфическая мутация 35delG объясняет большинство GJB2 мутаций, выявляемых в популяциях белых и представляет собой одну из наиболее частых мутаций, вызывающих болезнь (26, 31). Мутация 35delG представляет собой делецию guanine (G) в последовательности из 6 Gs в положении 30-35, что ведет к сдвигу рамки считывания и образованию преждевременного стоп кодона в нуклеотиде 38 (25, 26). Частота носительства 35delG мутации составляет 3.5-4.0% по данным Итальянских и Греческих популяций, это приводит к тому, что гомозиготность по этой мутации д. составлять 1 на 2500 новорожденных в этих популяциях (31, 32). Частота носителей мутации 35delG в южной Европе и регионе Средиземноморья выше, чем частота носительства главной ΔF508 мутации в гене CFTR, вызывающей кистозный фиброз (33). Градиент с юга на север по частоте носительства мутации 35delGописан среди Европейских стран со средней частотой носительства 2.8% в южной Европе до 1.3% в центральной и северной Европе (34). Первоначально было предположено, что высокое превалирование этой мутации обусловлено мутационной горячей точкой, но гаплотип, принимающий участие в очень небольшом хромосомном интервале у пациентов, гомозиготных по мутации 35delG указывает на то, что мутация происходит от общего родоначального основателя примерно 500 поколений и 10,000 лет тому назад (35). Вклад гена GJB2 и мутации 35delG в pre-lingual глухоту в разных исследованиях, базирующихся на изучении популяций представлен в Table 3. Относительный вклад гена GJB2 варьирует от 0 до 40% в изученных популяциях, демонстрируя генетическую гетерогенность, но некоторые исследования, базирующиеся на небольших количествах пациентов, а также на использовании др. критериев и методов скрининга мутаций, дают различающиеся данные.
Высокие частоты мутаций GJB2 иных, чем 35delG описаны в др. этнических группах, включая 167delT мутацию у евреев Ashkenazi (36), 235delC у японцев (37) и R143W у африканцев (38), подтверждая эффект основателя (36, 39).В целом около 90 различных мутаций GJB2 было описано в связи с рецессивной не-синдромальной потерей слуха (Ballana E et al. Connexins and Deafness. http://www.crg.es/deafness). Установлены генотип-фенотипические корреляции, которые демонстрируют, что пациенты с укорачивающими мутациями имеют значительно более тяжелые нарушения слуха, чем truncating/missense компаундные гетерозиготы и что пациенты с двумя missense мутациями имеют менее выраженную потерю слуха (40, 41).
В то время как ген GJB2 является главным геном, ответственным за не-синдромальную рецессивную глухоту во многих популяциях, имеются некоторые противоречия относительно роли GJB2 в доминантной глухоте(DFNA3) (42). Некоторые гетерозиготные GJB2 мутации, локализующиеся в определенном домене белка (первый внеклеточный домен), как установлено, сегрегируют с аутосомно-доминантной потерей слуха в небольшом количестве семей с разными фенотипами, представляющими pre-lingual с началом в поздном детской возрасте, от слабой до выраженной прогрессирующую потерю слуха (14, 43-45).
Мутации в гене
GJB2 ответственны также за синдромальные формы потери слуха, включая аутосомно-доминантную mutilating keratoderma с нейросенсорной глухотой (Vohwinkel синдром), др. формы аутосомно-доминантной palmoplantar keratoderma с глухотой и ectodermal dysplasia keratitis-ichtyosis-deafness синдром (46-50). Большинство из этих
GJB2 мутаций располагаются в первом внеклеточном домене (14). GJB2 является, следовательно, примером connexin гена (подобно GJB3 и GJB6), ассоциированного с не-синдромальной глухотой.
GJB6 (connexin 30) - DFNB1
Изучение 422 неродственных субъектов из Испании и Кубы с pre-lingual, не-синдромной глухотой выявило биаллельные GJB2 мутации у 129 пациентов (30.6%), тогда как 44 пациента (10.4%) имели мутацию только в одном GJB2 аллеле (51). У 22 из 44 субъектов, выявлена компаундная гетерозиготность по GJB2 мутации и по 342-kb делеции, укорачивающей ген GJB6 (51). Делеция, затрагивающая ген GJB6 является теломерной по сравнению с GJB2, она идентифицирована также у пациентов из семей евреев Ashkenazi и французов, которые были гетерозиготны по GJB2 мутации в транс положении по отношению к делеции GJB6 (52, 53). Исследования в 9 странах (54) показали, что делеция присутствует с высокими частотами в Испании, Франции, Израиле и Великобритании (5.9-9.7% от всех DFNB1 аллелей). Анализ гаплотипов показал, четкий эффект основателя для этой мутации у евреев Ashkenazi и некоторых странах западной Европы (54). Делеция пока не обнаружена в Турции, Италии и Австрии (54-56).
Ген GJB6, кодирующий connexin 30, расположен рядом с геном connexin 26, a делеция распространяется от 5' prime конца гена GJB6 в направлении гена GJB2. Следовательно, возможно, что делеция GJB6 делетирует также контрольные регионы гена GJB2. Значительно больше глухих индивидов гетерозиготны по одной GJB2 мутации, чем слышащих индивидов, что указывает на существование одной или более неизвестных GJB2 мутаций в некодирующей области GJB2, возможно с вовлечением в делецию некоторой контрольной области. Альтернативно, двугенное наследование также возможно, т.к. оба гена экспрессируются в одних и тех же структурах внутреннего уха и обладают 77% идентичностью аминокислотных последовательностей (57, 58). Более того, гомозиготные Gjb6-дефицитные мыши лишены endocochlear потенциала и обнаруживают дегенерацию кохлеарного сенсорного эпителия за счет клеточного апоптоза (59). Двугенное наследование описано для некоторых др. рецессивных нарушений, таких как retinitis pigmentosa и синдром Bardet-Biedl (60, 61) и оно было предположено в качестве механизма для немногих др. семей с глухотой, исходя из анализа сцепления (62, 63). Вторая в 232-kb делеция в локусе DFNB1 была описана недавно (64). Делеция была также обнаружена в транс положении с патогенными мутациями GJB2 у затронутых субъектов и возникала за счет неравной гомологичной рекомбинации (64).
Только одна доминантная
GJB6 мутация ассоциирована с не-синдромальной глухотой у небольшого количества пациентов (64). Как и в случае некоторых др. коннексинов, ограниченное количество
GJB6 missense мутаций вызывает врожденные аутосомно-доминантные нарушения кожи, hidrotic ectodermal dysplasia (Clouston синдром), который иногда ассоциирует с потерей слуха (66).
MYO7A (myosin VIIA) - DFNB2
Локус DFNB2 картирован с помощью геномных исследований в 11q13.5 в очень близко родственной семье из Туниса с сегрегирующей не-синдромальной, полной глухотой (67). Ген USH1B ответственен за аутосомно-рецессивную форму синдрома Usher типа 1 был выявлен в той же самой области хромосомы, а ответственным геном оказался MYO7A, кодирующий myosin VIIA (68). Аутосомно-рецессиваня глухота shaker-1 у мышей (обнаруживающих гиперактивность, мотание головой и кружение, обусловленные vestibular дисфункцией вместе с дисфункцией и прогрессивной дегенерацией Кортиева органа) также, как было установлено, обусловлена мутациями в гене myosin VII (69). Наконец. в 1997, MYO7A мутации были выявлены в 2-х из 8 Китайских ядерных семей с не-синдромальной, врожденной, полной потерей слуха и вестибулярной дисфункцией (70), и в исходной DFNB2-affected единокровной семье из Туниса (67, 71). Ген MYO7A является вторым DFNB геном, ассоциированным с рецессивной не-синдромальной глухотой (Table 1).
Myosins являются семейством базирующихся на актине молекулярных моторов, которые используют энергию от гидролиза АТФ, чтобы генерировать механическую силу. Неконвенционные миозины обладают функциями, которые не очень хорошо понятны, но как полагают, регулируют внутриклеточный мембранный транспорт. Они являются базирующимися на актине моторными молекулами, которые превращают химическую энергию в силу, позволяющую им перемещаться вдоль актиновых филамент (72, 73). Все миозины обладают общей структурной организацией, представленной законсервированным NH2-терминальным моторным доменом, сопровождаемым варьирующим количеством мотивов связывания легкой цепи (IQ) и очень отличающимся хвостом. Ген
MYO7A является типичным нестандартным миозином, состоящим из 48 кодирующих экзонов (72). Его экспрессия обнаруживается в нескольких тканях мышей и человека, включая сетчатку и улитку (68, 72).Во внутреннем ухе эмбрионов мышей только кохлеарные и вестибулярные сенсорные волосковые клетки экспрессируют ген myosin VIIA, указывая тем самым, что вестибулярная дисфункция и глухота могут быть результатом дефектного морфогенеза стереоцилий волосковых клеток, высоко специфические механические свойства которых являются критическими для процесса механотрансдукции (72). Более 50 самостоятельных
MYO7A мутаций было описано для USH1B, 4 разные мутации были найдены при DFNB2 и две при доминантной глухоте (DFNA11) (74-76). Мутации были разбросаны по всему гену
MYO7A (77).
MYO15 (myosin XV) - DFNB3
В этническом изоляте на Bali, 2% жителей имеют полную, врожденную, не-синдромальную, сенсоронейральную глухоту, наследуемую по аутосомно-рецессивному способу (78). С помощью картирования гомозиготности в этой семье ген DFNB3 был картирован в хромосоме 17p (79), это было подтверждено в Indian семье (80). На базе законсервированной хромосомной синтении аутосомно-рецессивная глухота у мышей shaker-2 была предположена гомологичной DFNB3 (80). Мыши shaker-2 имели мутацию в гене Myo15, в гене нестандартного миозина (81). Ген MYO15 человека был идентифицирован и картирован в DFNB3 критической области, а анализ последовательностей MYO15 выявил мутации в трех родственных семьях (82). Из общего количества более 100 состящих в кровном родстве семей сегрегация рецессивной потери слуха из Пакистана и Индии 7 семей обнаруживали сцепление с DFNB3 (83). Секвенирование гена MYO15 выявили 3 новых гомозиготных мутации, наследуемые в трех пакистанских семьях, подтвердившие, что мутации MYO15 ответственны за по крайней мере 5% рецессивной, полной потери слуха в этой популяции (83).
Полной длины миозин XV человека кодируется 66 экзонами (84), a исследования экспрессии продемонстрировали, что
MYO15 экспрессируется в ряде тканей помимо внутреннего уха (82). У мышей
shaker-2 выявлено присутствие очень коротких стереоцилий и длинные аномальные актин-содержащие структуры, которые проецируются от основания слуховой волосковой клетки, это указывает на то, что myosin XV необходим для организации актина в волосковых клетках (81, 85).
SLC26A4 (pendrin) - DFNB4
Геномные исследования в семье друзов (Druze) из Израиля с pre-lingual, тяжелой глухотой найдено сцепление с областью в 5-cM на хромосоме 7q31 человека в интервале, содержащем ген, мутантный при синдроме Pendred (SLC26A4) (86). Затронутые члены этой семьи, как позднее было установлено, имеют зоб (goitre) и синдром Pendred и, следовательно, эта семья не представляет DFNB (87). Крупная единокровная семья из Индии с врожденной, полной не-синдромальной глухотой обнаруживала сцепление с той же самой областью (DFNB4) (87, 88). Затронутые индивиды были гомозиготными по missense мутации, затрагивающей законсервированный остаток в SLC26A4, представляя др. пример, когда аллельные варианты ассоциируют как синдромальной, так и не-синдромальной формами глухоты (87). SLC26A4 мутации были выявлены в высокой пропорции индивидов с нейросенсорной потерей слуха и аномалиями височной кости от расширенного вестибулярного акведукта до дисплазии Mondini (89-92). В большой серии pre-lingually глухих пробандов (n = 374) и в семьях с кровным сродством (n = 318) из зап. и южной Азии, SLC26A4 мутации были обнаружены приблизительно в 5% случаев, что указывает на превалирование SLC26A4-ассоциированной глухоты в этих популяциях (93).
SLC26A4 кодирует трансмембранный белок pendrin, который функционирует как транспортер chloride и iodide и экспрессируется в щитовидной железе, внутреннем ухе и почках (94). Функциональные исследования показали, что мутации, ассоциированные с синдромом Pendred, характеризуются полной потерей транспорта chloride и iodide, тогда как мутантные аллели у пациентов с DFNB4 способны транспортировать и iodide и chloride, хотя и на значительно более низком уровне, чем pendrin дикого типа (90). Чтобы объяснить ассоциированные аномалии височной кости, было предположено, что SLC26A4 контролирует гомеостаз жидкости в мембранозном лабиринте, что в свою очередь влияет на развитие костного лабиринта (91).
Enlarged vestibular aqueduct (EVA) является частым симптомом у пациентов с синдромом Pendred (и при некоторых джр. синдромах, таких каксиндром BOR), но он может также присутствовать и как изолированная находка вместе с нейросенсорной потерей слуха. В большинстве случаев, были идентифицированы одна или две мутации
SLC26A4 (95, 96).
TMIE (transmembrane inner ear expressed gene) - DFNB6
Гомозиготность по descent была продемонстрирована в кровнородственной ядерной семье из южной Индии с pre-lingual глухотой, определяемой FNB6 локусом (97). 4 дополнительные небольшие семьи (одна индийская и три пакистантские) также обнаружили сцепление с DFNB6 (98). На базе законсервированной синтении между дистальной частью мышиной хромосомы 9 и хромосомы человека 3p, было предположено, что spinner линия глухих мышей может служить мышиной моделью потери слуха DFNB6 у людей (97). Мутации в новом гене Tmie, как было установлено, ответственны за потерю слуха и вестибулярную дисфункцию у мышей spinner (99), а затем и гомозиготные мутации в ортологе человека TMIE были выявлены у 5 DFNB6 семей (98).
Предполагаемый белок TMIE не обладает существенным сходством с каким-либо известным белком, а его экспресия продемонстрирована во многих тканях человека (98). Ультраструктура улитки у
spinner мышей обнаруживает нерегулярные пучки стереоцилий на апикальных поверхностях как внутренних, так и наружных волосковых клеток, это подтверждает участие Tmie в созревании волосковых клеток, но точная клеточная локализация и функция белка ещё не изучены (99).
TMC1 (transmembrane cochlear-expressed gene 1) - DFNB7
Локус для pre-lingual, тяжелой до полной потери слуха картирован в хромосоме 9q13-q21 в двух кровно-родственных семьях из Индии, определен как DFNB7 локус (100). Приблизительно 230 кровно-родственных индийских и пакистанских семей, передающих по наследству рецессивную, тяжелую до полной, pre-lingual глухоту, были скринированы на сцепление с DFNB7 регионом с подтверждением в 10 семьях (101). Новый ген, названный TMC1, идентифицирован в критическом интервале и 7 разных мутаций TMC1 было идентифицировано в 11 рецессивных семьях (в одной из двух original индийских семей DFNB7 и в 10 Indian/Pakistani семьях) (101). Мутации включали nonsense, frameshift, missense, геномные делеции и мутации сплайс-сайтов, все в гомозиготном состоянии. Мутации TMC1 кажутся довольно распространенной причиной рецессивной глухоты в Индии и Пакистане, но найдены ткже в семьях из Турции (102). Гетерозиготные missense мутации в TMC1 были описаны также в большой северо-американской семье с наследуемой аутосомно-доминантной, post-lingual, быстро прогрессирующей потерей слуха (DFNA36) (101).
Делеция в 1.6-kb, соотв. экзону 14 гена Tmc1 была выявлена при рецессивной глухоте у мутантных (dn) мышей (101, 103).
Ген
TMC1, как было установлено, принадлежит семейству трансмембранных channel-like (TMC) генов с 8 паралогами (TMC1-TMC8), которые предположительно кодируют белки с 6-10 трансмембранными доменами и новым законсервированной в 120-аминокислот последовательностью, названной TMC доменом (104). Гены
TMC6 и TMC8 идентичны генам
EVER1 и EVER2, мутантным при epidermodysplasia verruciformis, редком рецессивном генодерматозе, характеризующимся чувствитеьрностью к кожной инфекции к papilloma virus и ассоциированным не-меланомным раком кожи (104, 105). Анализ экспрессии
TMC1 выявил транскрипты в улитке плодов человека и у мышей во во внутренних и наружных слоуховых волосковых клетках, а также в нейросенсорном эпителии vestibular end organs (101).
TMPRSS3 (transmembrane serine protease) - DFNB8/DFNB10
Крупная кровно родственная пакистанская семья обнаруживает потерю слуха с началом в детстве и быстрым прогрессированием до полной глухоты к 4-5 годам, которая оказалась сцеплена с хромосомой 21q22.3, и определена как локус DFNB8 (106, 107). Независимый геномный поиск в крупной палестинской семье с тяжелой, pre-lingual глухотой также выявил сцепление с теломерной областью хромосомы 21 (DFNB10) (108, 109). Прямое секвенирование TMPRSS3 гена (transmembrane protease, serine 3), нового гена внутри критической области (110), выявило гомозиготную мутацию сплайс-сайта, приводящей к сдвигу рамки считывания в пакистанской DFNB8 семье (111). Палестинская DFNB10 семья, как было установлено, имеет сложную перестройку с делецией 8 bp и инсерцией 18 полных β-satellite повторяющихся мономеров в экзоне 11 гена TMPRSS3. Анализ сцепления в 159 кровно-родственных пакистанских семьях, передающих по наследству полную врожденную глухоту, выявил 5 семей с потенциальным сцеплением с DFNB8/DFNB10 локусом (112). Гомозиготные TMPRSS3 мутации были выявлены в 4 из этих семей (112). Секвенирование TMPRSS3 у 64 глухих северо-американских пациентов (как из семейных, так и спорадических случаев) не выявило каких-либо мутаций (112). Две гомозиготные missense мутации в serine protease домене были выявлены в двух из 39 изученных кровно-родственных семей из Туниса с тяжелой до полной глухоты (113). Крупное исследование 448 неродственных белых пациентов с тяжелой полной глухотой в детстве из Греции, Испании, Италии и Австрии идентифицировали только 4 мутации, указывающие на то, что мутации TMPRSS3 не являются частой причиной pre-lingual глухоты в популяциях белых(114).
Ген
TMPRSS3 имеет 13 экзонов, кодирующих transmembrane (TM), low-density-lipoprotein receptor A (LDLRA), scavenger-receptor cysteine-rich (SRCR) и serine protease домены, сходные с др. протеазами (111). Мышиный ортолог
TMPRSS3 экспрессируется в спиральном ганглии, в поддерживающих клетках Кортиева органа и в stria vascularis, преимущественно в мембранах endoplasmic reticulum (115). Эпителиальный натриевый канал EnaC, который участвует в регуляции концентрации натрия в эндолимфе, обнаруживает сходную с Tmprss3 экспрессию во внутреннем ухе крыс. Поэтому было предположено, что EnaC является субстратом для TMPRSS3 (115). Исследования функциональной экспрессии в ооцитах
Xenopus laevis продемонстрировали, что TMPRSS3 существенно активирует ENaC, в то время как
TMPRSS3 missense мутации вызывают DFNB8/DFNB10 глухоту из-за неспособности активировать ENaC в этой модели (115).
OTOF (otoferlin) - DFNB9
Ген, ответственный за pre-lingual, полную, нейросенсорную глухоту был картирован в результате поиска по всему геному в хромосоме 2p23-p22 (DFNB9 локус) в сунитской кровно-родственной семье, живущей изолированно в селении Ливана (116). Кровно-родственные семьи из восточной Турции (117) и три (из 30) ливанские семьи (118) также обнаруживали локус DFNB9. Гомозиготная мутация по новому гену (OTOF), кодирующему otoferlin, выявлена в 4-х ливанских семьях (118). Скрининг мутаций в кровно-родственной семье из Индии с тяжелой до полной, pre-lingual, нейросенсорной потерей слуха выявил гомозиготную мутацию сплайс-сайта (119). Др. OTOF мутации были описаны в одиночных семьях, включая мутацию сплайс-сайта в кровно-родственной семье Druze из Галилеи (120), nonsense мутацию в кровно-родственной семье из Объединенных Арабских Эмиратов (121), две missense мутации в кровно-родственной турецкой семье, ранее картировано по DFNB9 локусу (117, 122) и мутацию преждевременного стоп кодона в экзоне 22 (Q829X мутация) в Испанской семье (123). 11 случаев (10 в Испании и 1 на Кубе) из 269 неродственных случаев рецессивной потери слуха (негативных в отношении мутации GJB2) также имели Q829X мутацию (123). Анализ гаплотипов строго подтвердил происхождение из общего родоначальника для мутации Q829X, которая, по-видимому, ответственна за примерно 3% от всех случаев рецессивной, pre-lingual глухоты в Испанских популяциях (123).
OTOF мутации были также описаны при несиндромальной, рецессивной auditory neuropathy, которая характеризуется от умеренной до полной, сенсоронейральной потери слуха с нормальной otoacoustic emissions (OAEs), указывающей на сохранность функции наружных волосковых клеток и отсутствие какой-либо др. обнаруживаемой периферической нейропатии и с отсутствием пользы от слухового аппарата (124, 125). В крупном исследовании Испанских пациентов с OTOF-ассоциированной глухотой, 11 из 21 субъектов, несущих две мутации в гене OTOF, имели сохранной OAEs, по крайней мере, в одном ухе (126). Следовательно, генетический диагноз пациентов с полным нарушением слуха, но с сохранением OAEs, д. склоняться к гену OTOF. Более того, эти находки важны для программ скрининга новорожденных в отношении нарушения слуха с использованием OAEs в качестве первого теста детекции (126).
Ген
OTOF человека, как было установлено, состоит из 48 кодирующих экзонов, дающих белок в 1997 аминокислот otoferlin с альтернативно сплайсируемыми транскриптами, дающими несколько длинных изофром (с 6 C2 доменами) и короткие изоформы (с 3 C2 доменами) (119). Анализ otoferlin выявил трансмембранный домен на С-конце с остальной чатью белка, предположительно располагающейся в цитоплазме и тремя Ca
2+-связывающими C2 доменами (118). Предполагается, что Ca
2+ запускает слияние мембран синаптических пузырьков (118). Обнаружена гомология с фактором сперматогенеза
Caenorhabditis elegans fer-1 (127), с dysferlin человека (кодируемого
DYSF), скелетно-мышечным геном, мутантным при Miyoshi myopathy и при limb-girdle muscular dystrophy type 2B (128, 129), и с myoferlin человека (кодируемого
MYOF) (130). Строгая экспрессия в тканях мышей наблюдалась в улитке, vestibule и головном мозге (118).
In situ гибридизация во внутреннем ухе мышей выявила преимущественную экспрессию во внутренних волосковых клетках улитки и вестибулярном нейроэпителии.
CDH23 (otocadherin) - DFNB12
Анализ сцепления в кровно-родственной сунитской семье, проживающей в деревне в Сирии, выявил локус DFNB12 для pre-lingual, полной нейросенсорной тугоухости на хромосоме 10q21-q22 (131) в интервале, перекрывающемся с Usher syndrome type 1D (USH1D) (132). 13 кровно-родственных семей (из Пакистана, Индии и Турции) передают по наследству аутосомно-рецессивную, полную, врожденную глухоту (n = 7) или фенотипы USH1 (n = 6), демонстрируя сцепление с 10q21-q22 (133). Секвенирование всех 18 генов кандидатов продемонстрировало мутации в новом cadherin-подобном гене CDH23 в 9 из 13 семей (5 DFNB12 семей и 4 USH1D семей) (133). 2 мут ации были также идентифицированы в крупной, инбредной кубинской USH1 семье (134), мутации CDH23 объясняют примерно 10% пациентов в исследовании 52 случаев USH1 (135). В крупном исследовани, включающем 69 семей с синдромом Usher типа 1D и 38 семей с рецессивной, не-сидромальной глухотой из разных стран, всего было идентифицировано 36 разных мутаций CDH23 в 45 семьях (136). 9 из 38 семей с рецессивной, не-синдромальной глухотой, извлеченных из исходной популяции с 157 пациентами не-синдромальной глухотой имели мутацию CDH23, указывая тем самым, что до 5% не-синдромальной глухоты вызывается мутациями в гене CDH23 (136). Четкая генотип-фенотип корреляыция установлена в этом исследовании: все Usher пациенты за исключением 3-х, представленных как атипические фенотипы Usher синдрома, обнаруживают одну или две укорачивающие мутации, в то время как все DFNB12 пациенты обнаруживают две missense мутации(136). DFNB12 фенотип демонстрирует значительную внутри- и межсемейную вариацию с потерей слуха, варьирующей от умеренной до полной глухоты и возрастом установки диагноза между 3 мес. и 6 годами (136). Не отмечено нарушений полей зрения, офтальмологические проверки продемонстрировали безсимптомные retinitis pigmentosa-подобные проявления (subnormal ERG response, subnormal fundus examination) (136).
Мутации потери функции у мышей в ортологе Cdh23 вызывают дезорганизацию стереоцилий во внутренних и наружных волосковых клетках у мышей waltzer, это указывает на то, что otocadherin является критическим компонентом для собственно формирования пучков волосков.
Ген
CDH23 является очень крупным геном, состоящим из, по крайней мере, 70 экзонов, кодирующих 3353 аминокислот. Northern blot анализ CDH23 выявил 9.5-kb транскрипт, экспрессирующийся прежде всего в сетчатке, экспрессия в улитке была также продемонстрирована, но с помощью секвенирования PCR продуктов из кДНК библиотеки улитки человека (133). Ген
CDH23 принадлежит к сверхсемейству cadherin белков межклеточной адгезии, которые обычно имеют крупные внеклеточные домены (характеризующиеся cadherin повторами, которые, как было показано, обеспечивают межклеточную адгезию), пронизывающую мембрану область и цитоплазматические домены, высоко дивергентные среди членов семейства (137). Данные на мышах выявили участие cadherin 23, harmonin (see DFNB18) и myosin VIIa (see DFNB2) в одной функциональной сети, существенной для гарантии слипчивости стереоцилий из пучков волосков (138, 139). CDH23, как было показано, является частью верхушечных связей, участвующих в поперечном связывании стереоцилий (140, 141).
STRC (stereocilin) - DFNB16
Анализ сцепления 29 пакистанских и 12 семей с Ближнего Востока показал, что 3 кровно-родственных семьи обнаруживают сцепление с хромосомой 15q15-q21, обозначенной как DFNB16 локус для не-синдромной, аутосомно-рецессивной глухоты (142). Позднее то же самое сцепление выявлено в большой кровно-родственной испанской семье (143). С использованием кДНК библиотеки внутреннего уха мышей, был выделен новый ген STRC, экспрессирующийся почти исключительно во внутреннем ухе и совпадающий с некоторыми геномными клонами из DFNB16 хромосомной области человека (144). Скрининг мутаций в уже описанных семьях (142, 143) выявил две мутации сдвига рамки считывания и крупную делецию в двух из семей (144). Переоценка области гомозиготности в двух др. семьях с DFNB16подтвердила, что DFNB16 локус может содержать второй ген глухоты (144).
Ген
STRC содержит 29 кодирующих экзонов и, как было установлено, является тандемно удвоенным со стоп кодоном в 20 экзоне в копии B copy, которая может быть псевдогеном (144). Предполагаемый белок stereocilin не обнаруживает существенной гомологии с др. известными белками (144). Иммунофлюоресцентные исследования показали, что во внутреннем ухе мышей stereocilin экспрессируется только в сенсорных волосковых клетках с интенсивным окрашиванием пучков волосков, составляющих стереоцилии (144).
USH1C (harmonin) - DFNB18
Pre-lingual, полная, не-синдромальная, нейросенсорная глухота наследующаяся в крупной, кровно-родственной индийской семье, была картирована с помощью поиска по всему геному в хромосоме 11p15.1-p14 (DFNB18) (145), соответ. области Usher синдрома типа 1C (USH1C) (146). Мутация сплайс-сайта в интроне 12 гена USH1C обнаружена в гомозиготном состоянии в индийской семье (147). Дополнительные мутации были идентифицированы позденее в др. семьях (148-150). Harmonin очень редко дает не-синдромальную глухоту, т.к. скрининг мутаций в 32 китайских семьях с несиндромной, pre-lingual глухотой (150) и в 16 парах сибсов белых из Великобритании (151) выявило только 1 мутацию.
Ген USH1C содержит 28 экзонов (147) и кодирует PDZ домен-содержащий белок, harmonin (от греческого слова armonia, означающего 'assembling in a correct order'). Иммуногистофлюоресценция выявляет harmonin в сенсорной области внутреннего уха, особенно в цитоплазме и стереоцилиях волосковых клеток (147). Идентифицировано 8 разных транскриптов во внутреннем ухе мышей (147). Harmonin, как было установлено, соединяется с otocadherin (see DFNB12) и взаимодействует с myosin VIIA (see DFNB2), указывая на функциональную единицу, лежащую в основе образования связанного пучка волосковых клеток (138, 139).
Мутации в мышином гене ортологе
Ush1c вызывают врожденную глухоту и тяжелые нарушения баланса (deaf circler) (152). Патология внутреннего уха мутантных
deaf circler мышей указыает на прогрессирующую потерю волосковых клеток и вторичную дегенерацию клеток спирального ганглия (152). Дегенерации волосковых клеток предшествует дезорганизация стереоцилий, видимая в ЭМ (152).
TECTA (α-tectorin) - DFNB21
Аназиз сцепления в кровно-родсвенной ливанской семье с pre-lingual, от тяжелой до полной потери слуха выявил новый локус для рецессивной глухоты DFNB21 (153) в области хромосомы 11q23-q25, соотв. гену TECTA , ответственному за DFNA8/DFNA12 глухоту (154-156). В семье с DFNB21 выявлена мутация сплайс-сайта в интроне 9 TECTA (153). Гомозиготные мутации сдвига рамки считывания в гене TECTA были нацдены в двух кровно-родственных семьях из Ирана и Пакистана (157). Мыши, гомозиготные по делеции в α-tectorin, имеют tectorial мембраны, которые отслоены от кохлеарного эпителия и лишены всего не-коллагенового матрикса (158). Доминантные TECTA missense мутации описаны в DFNA8/DFNA12 семьях Австрии, Бельгии, Франции и Швеции (156, 159, 160). Эти missense мутации, по-видимому, имеют доминантно-негативный эффект, который нарушает структуру текториальной мембраны.
TECTA кодирует α-tectorin, один из основных не-коллагеновых компонентов внеклеточного матрикса текториальной мембраны, которая контактирует с пучками стереоцилий сенсорных волосковых клеток (158).
OTOA (otoancorin) - DFNB22
Мышиный ген
otoancorin выделен из кДНК библиотеки из сенсорного эпителия вестибулярного аппарата мышей , он предположительно кодирует закрепленный в мембране белок со специфической для внутреннего уха экспрессией (161). Не выявлено гомологии с доменами известных белков и установлена слабая гомология последовательностей с megakaryocyte potentiating factor (MPF)/mesothelin предшественником, это указывает на то, что otoancorin может действовать как адгезивная молекула (161, 162). Otoancorin во внутреннем ухе мышей специфически локализуется на апикальной поверхности клеток, с помощьюкоторой они контактируют с лежащим поверх бесклеточным гелем (161). Otoancorin, как полагают, обеспечивает прикрепление текториальной мембраны в улитке и otoconial мембраны и cupulae в vestibule. Соотв. ген человека,
OTOA, состоит из 28 экзонов и картируется в хромосоме 16p12.2 (161). Одна кровно-родственная палестинская семья с умеренной до тяжёлой, pre-lingual, нейросенсорной рецессивной глухотой из коллекции в 200 глухих семей, обнаружила наследование, связанное с интервалом 16p13.1-q11.2 (DFNB22), хромосомной областью, содержащей OTOA (161). Мутация сплайс-сайта в соединении exon 12/intron 12 ко-сегрегировала с нарушением слуха в этой семье (161). Не обнаружено др.
OTOA мутаций у 150 пробандов из семей с не-синдромальной, рецессивной глухотой (в основном белых и происходящих из Китая) и не найдено др. семей с глухотой, сцепленных с локусом DFNB22 в дополнительной коллекции из 150 крупных семей (из Израиля и Испании) (161). Это указывает на то, что мутации
OTOA не являются частой причиной глухоты.
PCDH15 (protocadherin 15) - DFNB23
Ген
PCDH15, кодирующий protocadherin 15, ранее был признан ответственным за Usher синдром типа 1F (163, 164), в то время как рецессивные мутации
Pcdh15 вызывают глухоту у Ames waltzer (
av) мышей (165). Ген
PCDH15 является. следовательно, прекрасным кандидатом для не-синдромальной наследственной глухоты. Полсе скрининга 400 семей с аутосомно-рецессивной, pre-lingual потерей слуха, две семьи оказались сцепленными с PCDH15 регионом (DFNB23), анализ последовательностей продемонстрировал гомозиготность по PCDH15 missense мутациям в двух семьях (166). Иммунореактивность protocadherin 15 была выявлена у мышей в фоторецепторах сетчатки, Кортиевом органе и вестибулярных волосковых клетках. Иммунореактивность обнаруживалась вдоль длины стереоцилий, в cuticular пластинке и диффузно распределена в цитоплазме внутренних и наружных волосковых клеток (166).
Ames waltzer (av) мыши обнаруживают дезорганизованные пучки стереоцилий и дегенерацию нейроэпителия внутреннего уха (165).
CLDN14 (claudin 14) - DFNB29
Локус DFNB29 был картирован на хромосоме 21q22.1 путем анализа сцепления в двух крцпных кровно-родственных пакистанских семьях с pre-lingual, полной глухотой (167). Критический интервал содержит ген CLDN14, который является прекрасным кандидатом. Анализ последовательностей CLDN14 выявил гомозиготную делецию одиночного нуклеотида в трансмембранном домене 2 в двух DFNB29 семьях (167). Среди 100 пакистанских семей с рецессивной глухотой только эти две семьи обнаруживали сцепление с CLDN14 (167).
CLDN14 мутаций не было выявлено при изучении 60 турецких семей с аутосомно-рецессивной, не-синдромальной потерей слуха (56).
Cldn14 нокаутные мыши, полученные за счет целенаправленной делеции в
Cldn14, обнаруживали нормальный endocochlear потенциал, но глухота была обусловлена быстрой дегенерацией наружных волосковых клеток улитки, что сопровождалось более медленной дегенерацией внутренних волосковых клеток (168). Claudins представляют собой мультигенное семейство интегральных белков мембран, идентифицированных как как крупные адгезивные молекулы, действующие в межклеточных плотных соединениях (169). Иммунофлюоресцентные исследования у мышей на постнатальный день 4 продемонстрировали экспрессию claudin 14 в области внутренних и наружных волосковых клеток Кортиева органа и в сенсорном эпителии вестибулярных органов (167). Между постнатальным днем 4 и 8 экспрессия claudin 14 снижается в волосковых клетках ипоявляется в поддерживающих клетках Кортиева органа, паттерн экспрессии совпадающий с развитием endocochlear потенциала (167). Было предположено, что отсутствие claudin 14 в плотных соединениях в Кортиевом органе ведет к изменению ионной проницаемости околоклеточного барьера ретикулярной ламины и что продолжительное воздействие на базо-латеральные мембраны наружных волосковых клеток высоких концентраций калия может быть причиной клеточной гибели волосковых клеток (168).
MYO3A (myosin IIIA) - DFNB30
Геномный поиск в трех поколениях израильской семьи с прогрессирующей потерей слуха предположил сцепление с хромосомой 10p, определяющей локус DFNB30 (170). Потеря слуха начинается во второй декаде жизни, сначала затрагиваются высокие частоты и к 50 годам наступает тяжелая потеря высоких и средних частот и умеренная потеря низких частот. MYO3A картирован в DFNB30 области и является прекрасным геном кандидатом (171), т.к. 4 др. миозина уже ассоциировали с потерей слуха (170). 3 разных мутации потери функции MYO3A , как было установлено ко-сегрегируют с потерей слуха в Израильской семье, 7 членов которой оказались гомозиготными и 11 компаундными гетерозиготами по парам мутантных аллелей. Не описано др. семей с мутациями MYO3A.
Myosin IIIA является актин-зависимым моторным белком, относящимся к классу III нестрандартных миозинов с 36% идентичностью с
Drosophila NINAC, мутации в котором вызывают дегенерацию сетчатки (171). Исходя из консервации доменов между NINAC и MYO3A, актин-связывающая функция, скорее всего, законсервирована, но лиганд из myosin IIIA хвостового домена предстоит идентифицировать (170). Т.к. моторный домен MYO3A пептида больше всего напоминает таковой myosin VIIA человека, то возможно, что MYO3A участвует в процессе механотрансдукции, но точная функция его в ушах млекопитающий пока неизвестна. Myosin IIIA экспрессируется в сетчатке человека (171), а экспрессия у мышей обнаруживается в улитке, где она ограничивается нейросенсорным эпителием, особенно внутренними и наружными волосковыми клетками (170).
WHRN (whirlin) - DFNB31
Картирование гомозиготности в кровно-родственной палестинской семье из Иордана выявило локус (DFNB31) для pre-lingual, полной потери слуха в хромосоме 9q32-q34 (172). Мышиная область, синтеничная DFNB31 интервалу на хромосоме 4, содержит локус для рецессивной глухоты у мутантов whirler (wi), который в гомозиготном состоянии у взрослых вызывает shaker-waltzer синдром: глухота и круговращение с потряхиваниями головы (172-175). Анализ последовательностей нового гена (Whrn кодирует whirlin) в этом регионе идентифицировал делецию в 592 bp у wi мышей (173), и гомозиготную nonsense мутацию в исходной DFNB31 семье (172). Скрининг 150 пробандов из семей с рецессивной глухотой (в основном Европейского и Китайского происхождения) по мутациям в WHRN не выявил каких-либо аномалий (173), в то время как анализ сцепления в 63 туниских семьях идентифицировал сцепление с DFNB31 только в одной семье (176), указывая тем самым, что WHRN мутации не вносят существенного вклада в рецессивную глухоту.
Ген человека представлен 12 экзонами с тремя PDZ доменами и одним доменом, богатым пролином, ближайшим к нему родственным белком является harmonin (see DFNB18), который также содержит три PDZ домена (173).
Иммунофлюоресцентные исследования у мышей выявили экспрессию whirlin, перекрывающуюся с окрашиванием актина в стереоцилиях на растущих концах актиновых филамент (173). Эти находки подтверждают, что whirlin действует, контролируя полимеризацию актина и рост мембран стереоцилий (173, 177). Удлинение стереоцилий, как было установлено, является дефектным у
wi гомозигот с дегенерацией в конечном счете как внутренних. так и наружных волосковых клеток (178).
ESPN (espin) - DFNB36
Локус DFNB36 был картирован при поиске по всему геному в двух кровно-родственных пакистанских семьях, наследующих pre-lingual, полную оптерю слуха и вестибулярную areflexia (179). Интервал сцепления на хромосоме 1p36.3 содержит ESPN (espin), ген, как известно, вызывающий глухоту и вестибулярную дисфункцию у мышей jerker (180). Две гомозиготные ESPN мутации сдвига рамки считывания выявлены в двух пакистанских семьях (179).
Ген
ESPN человека состоит из 13 экзонов и. по-видимому, кодирует белок в 854 аминокислоты с 8 ankyrin повторами, двумя областями, богатыми пролином, actin-связывающим WH2 доменом и суперскрученными доменом, важным для связывания актина в пучки (179). Espins являются белками, связывающими актин в пучки. В улитке и преддверии врнутреннего уха мышей espin локализуется в основном в стереоцилиях (180). Espin отсутствует в стереоцилиях мышей
jerker, приводя в конечном итоге к полной потере всех сенсорных волосковых клеток (180).
MYO6 (myosin VI) - DFNB37
Сканирование генома крупной кровно-родственной пакистанской семьи с полной, нейросенсорной, pre-lingual потерей слуха выявило ко-сегрегацию с 6q13 и обозначило DFNB37 локус (181). Помимо глухоты имеется вестибулярная дисфункция и умеренный лицевой дисморфизм в этой семье. Две пакистанские семьи с не-синдромальной глухотой позднее также оказались сцепленными с локусом DFNB37 (181). Область сцепления включает ген MYO6, кодирующий myosin VI. MYO6, как известно ответственен за аутосомно-доминантную форму post-lingual прогрессирующей глухоты в одной итальянской родословной (DFNA22) (182), тогда как мышиный Myo6 ответственен за глухоту и вестибулярную дисфункцию у Snell's waltzer (sv) мышей (183). Скрининг мутаций по гену MYO6 выявил гомозиготные мутации в трех пакистанских семьях (181). Missense мутация в высоко законсервированном моторном домене MYO6 оказалась ассоциированной с аутосомно-доминантной нейросенсорной потерей слуха ко-сегрегирующей с hypertrophic cardiomyopathy и удлинением QT интервала в одной родословной (184). Кардиальные симптомы были слабые или отсутствовали у большинства членов затронутых семей и могут быть пропущены в др. родословных с MYO6 глухотой (184).
Ген
MYO6 относится к нестандартным миозинам, экспрессирующимся на высоком уровне в основании стереоцилий и наружных и внутренних волосковых клетках (185). У
Snell's waltzer мышей видны гигантские стереоцилии с дегенерацией волосковых клеток (183).
COL11A2 (collagen 11α2) - DFNB53
Сканирование по всему геному в кровно-родственной семье из Ирана выявило новый локус на 6p21.3 (DFNB53) (186). Клинические находки соответствовали не-синдромальной, pre-lingual, полной потере слуха. Интервал сцепления содержит ген COL11A2, ассоциированный с DFNA13 доминантной, не-синдромальной глухотой в американских и датских семьях (187). Скрининг мутаций по гену COL11A2 в итальянской семье выявил missense мутацию в экзоне 21 в гомозиготном состоянии у затронутых индивидов (186).
Описано несколько др. мутаций
COL11A2, включая рецессивные мутации при otospondylomegaepiphyseal dysplasia (OSMED) (188) и доминантные мутации при non-ocular Stickler syndrome (189), все являются синдромными типами потери слуха. Генотип-фенотипические сравнения указывают на то, что тип и расположение мутации является критическим детерминантом в предопределении фенотипа COL11A2-ассоциированных болезней diseases (186).
GJB3 (connexin 31)
ген GJB3, кодирующий белок щелевых соединений connexin 31, был клонирован и картирован на хромосоме 1p35-p33 и, как полагают, является хорошим кандидатом для наследственного нарушения слуха (190). Сегодня ни один из известных DFNB локусов не был картирован в хромосомной области 1p35-p33. Скрининг мутаций в 25 китайских семьях с рецессивной глухотой выявил две небольшие семьи с ранним началом потери слуха с компаундной гетерозиготносттью по тем же самым двум GJB3 мутациям (191). Некоторые GJB3мутации с неизвестным значением были также описаны: гетерозиготная мутация GJB3 была идентифицирована в небольшой семье с двумя затронутыми детьми среди 60 Турецких семей с аутосомно-рецессивной, не-синдромальной потерей слуха, но мутация унаследована от отца, который не имел тугоухости (56). Скрининг мутаций среди 63 индивидов с не-синдромальной тугоухостью выявил гетерозиготную R32W мутацию у двух пациентов с поздним началом потери слуха (192), но эта мутация присутствовала и в контрольной популяции (193). GJB3 мутации оказались также ассоциированными с аутосомно-доминантной high-frequency потерей слуха (DFNA2) в двух небольших китайских семьях (190). Разные GJB3 мутации были также ответственны за аутосомно-доминатаные erythrokeratodermia variabilis (EKV) (194), в то время как рецессивная EKV в небольшой еврейской семье была обусловлена гомозиготной GJB3 missense мутацией (195). Наконец, 3-bp GJB3 делеция была обнаружена в испанской семье, наследующей аутосомно-доминантную, умеренную потерю слуха с периферической нейропатией (196).
Предполагаемый GJB3 белок connexin 31 обладает 4 гидрофобными трансмембранный домен-подобными мотивами, структурой, сходной с той, что имеется у др. коннексинов и обладает гомологией в 76% с GJB2 человека (21, 190). У взрослых крыс экспрессия GJB3 выявляется в кортексе, спинном мозге и внутреннем ухе(190).
GJA1 (connexin 43)
Мутации в гене
GJA1, кодирующем белок щелевых соединений connexin 43, в 6q21-q23.2 (197, 198) вызывают oculodentodigital dysplasia синдром (199), syndactyly type 3 и возможно heterotaxy (200). В связи с вовлечением др. коннексинов в глухоту, GJA1 также рассматривается как кандидат для не-синдромальной глухоты (201). Скрининг мутаций по гену GJA1 у 26 глухих пробандов африканцев американского происхождения с рецессивной или спорадической, pre-lingual, полной глухотой выявил гомозиготные missense мутации у 4-х пациентов (201). Однако, эти мутации в действительности затрагивают псевдоген connexin 43 на хромосоме 5, необходимы дополнительные данные, чтобы определить роль GJA1 в глухоте.
SLC26A5 (prestin)
Prestin принадлежит к solute carrier (SLC) семейству генов 26, которые кодируют белки, связанные с транспортерами анионов. Идентифицировано уже 9 генов SLC26A у человека, а мутации в SLC26A2, SLC26A3 и SLC26A4 генах ответственны за три самостоятельные рецессивные нарушения: diastrophic dysplasia, congenital chloride diarrhea и Pendred syndrome/DFNB4, соотв. (202, 203). Это делает prestin холрошим геном кандидатом для генетической глухоты. Скрининг мутаций у 220 белых пробандов с тугоухостью выявлена гомозиготная мутация сплайс сайта у двух глухих индивидов с тяжелой или полной, pre-lingual глухотой (204). 7 из 220 (3.2%) глухих пробанда были найдены гетерозиготными по той же самой мутации (204). Потеря слуха у этих гетерозиготных индивидов варьирует от умеренной до полной и с возрастом начала от рождения до 35 лет (204). Та же самая мутация найдена у одного гетерозиготного индивида среди 150 белых контрольной группы с нормальным слухом, но этот индивид не был протестирован аудиологически и мог иметь умеренную потерю слуха (204). Эти находки указывают на то, что эта мутация не полностью пенетрантна в гетерозиготном состоянии (204). Скрининг пациентов из семей DFNB14 и DFNB17 из Ливана и Индии, соотв., позволил картировать 7q31 вблизи гена SLC26A5 тогда как у 150 глухих пробандов из др. этнического фона не выявлено ни одной SLC26A5 мутации (204). Исследование на мышах с гомозиготной нокаутной по prestin мутацией выявило прогрессивную потерю наружных и внутренних волосковых клеток со снижением порога слышимости до 40-60 dB, в то время как гетерозиготные нокауты обнаруживали потерю слуха (205).
Ген
SLC26A5 человека на хромосоме 7q22.1 имеет 21 экзон, кодирующие белок prestin с сильно гидрофобной сердцевиной из 12 трансмембранных доменов с N- и C-окончаниями. локализованными в цитоплазме (204). Он специфически и на высоком уровне экспрессируется в наружных волосковых клетках, выстилающих латеральную стенку в виде тесно упакованных групп (204, 206). Он является главным белком наружных волосковых клеток улитки и предположительно участвует в амплификации вибраций в улитке, которая передается с помощью внутренних волосковых клеток (207).
ATP2B2 modifier gene
Потеря слуха, обусловленная мутациями в CDH23 или MYO6 может быть модифицирована с помощью гетерозиготности по гипофункциональному варианту в гене
ATP2B2, кодирующем plasma-membrane calcium pump PMCA2 (208), отражая взаимодействие между этими генами.
Conclusion
The first successful linkage study of an autosomal-recessive form of non-syndromic deafness was reported in 1994, and the first recessive deafness genes (GJB2 and MYO7A) were identified in 1997. A total of 23 different DFNB genes have been reported until today (December 2005). Several of the genes are involved in both recessive and dominant non-syndromic deafness (GJB2, GJB3, GJB6, MYO6, MYO7A, TMC1, TECTA, and COL11A2) or in both non-syndromic and syndromic deafness (GJB2, GJB3, GJB6, MYO7A, SLC26A4, CDH23, USH1C, PCDH15, COL11A2, and MYO6). The phenotype of non-syndromic, recessive deafness is usually severe to profound, pre-lingual, non-progressive deafness, in contrast to dominant hearing loss, which is usually post-lingual and progressive from mild to severe. Nearly all genes both for recessive and dominant non-syndromic forms cause sensorineural hearing loss. Most of the genes for recessive deafness have been identified by positional genetics in inbred multiplex families in ethnic isolates. Only a few of the genes including OTOA, PCDH15, GJB3, GJA1, and SLC26A5 have been found to be associated with recessive deafness by a functional candidate gene approach.
As many as 23 different DFNB proteins have been implicated in deafness to date, including proteins involved in the cytoskeleton such as the myosins, structural proteins such as the tectorin protein, ion transport proteins including several gap junction proteins and ion channels, and several genes with unknown function (Table 2).
Two important networks come into picture – a network of genes regulating potassium homeostasis in the cochlea and a network of genes forming stereocilia. Potassium homeostasis is very important in the cochlea, more precisely in repolarization and resetting of the hair cell potential. Initial depolarization of the hair cells is caused by influx of potassium ions from the endolymph through the tip links of the stereocilia. The hair cells are repolarized when the potassium ions leave these cells via potassium channels probably formed by the KCNQ4 gene and enter the supporting Deiter cells. They then diffuse through gap junctions formed by connexins to the stria vascularis. These potassium ions are secreted back into the endolymph through potassium channels formed by the KCNQ1 and KCNE1 gene products, thereby resetting the mechanoelectrical transduction system. Also, the pendrin gene SLC26A4, different gap junction genes encoding connexins (GJB2, GJB3, and GJB6), and the tight junction protein gene CLDN14 are involved in ion homeostasis in the cochlea and implicated in deafness.
The second network frequently implicated in deafness is located in the stereocilia and consists of the myosin VIIA, myosin XV, whirlin, harmonin, cadherin 23, and protocadherin 15 genes (209, 210). These proteins are located in the stereocilia and/or tip links connecting stereocilia, which are the mechano-electrical transducers transducing sound vibrations into electrical signals. Digenic inheritance with heterozygous mutations in two of these genes encoding cadherin 23 and protocadherin 15 has been demonstrated recently in recessive deafness both in mice and in humans (211).
Many of the genes have so far been reported in only single or a few consanguineous deafness families, and their contribution to deafness on a population base might therefore be limited or is currently unknown (212). Only the GJB2, GJB6, MYO15, SLC26A4, TMC1, OTOF, CDH23, and STRC genes have been implicated in a significant number of families (Table 4). Most of the deafness genes identified so far are large, with many exons and no mutational hotspots. Only the GJB2 gene is small with only one coding exon and a very prevalent mutation 35delG in Caucasians.
Due to this genetic heterogeneity, private nature of most mutations, and large size of most deafness genes, the molecular diagnostic possibilities in recessive deafness are limited apart from the mutation analysis of the GJB2 gene. However, it should be possible soon to develop an array, which detects most of the mutations previously described.
Сайт создан в системе
uCoz  and PJ Willemsb
and PJ Willemsb