Myeloproliferative disorders (MPD) это группа гематологических заболеваний, в которых первичное нарушение происходит на уровне мультипотентных гематопоэтических стволовых клеток, ведущее к повышенной продукции одного или более типов кровнях клеток. Три основных заболевания в группе polycythaemia vera (PV), essential thrombocythaemia (ET) и idiopathic myelofibrosis (IMF). PV характеризуется повышением эритроцитов, белых клеток и тромбоцитов и клинически проявляется полнокровием, зудом и спленомегалией. Заболевание может быть осложнено тромбоэмболическим феноменом и гемморагиями и на конечной стадии может прогрессировать в myelofibrosis ир острую лейкемию. ET характеризуется повышенным количеством тромбоцитов. Клинически оно часто безсимптомно, но тромбоэмболические события приводят к диагнозу. Имеется незначительная склонность к прогрессированию в myelofibrosis и острую лейкемию, на которые можно повлиять обещепринятым лечением. IMF определяется по leukoerythroblastic картине крови, спленомегалии и фиброзу костного мозга. Картина крови включает анемию, thrombocythaemia или thrombocytopenia и варьирующие количества белых клеток. Болезнь часто прогрессирует неумолимо в зависимую от трансфузий анемию, симптоматическую спленомегалию и трансформируется в острую лейкемию. Ряд др. биологических феноменов описан в гематопоэтических клетках от PV пациентов и также при др. MPDs, большинство из которых вызывает дисрегуляцию ключевых сигнальных медиаторов. Ключевые молекулярные события в патогенезе этих нарушений плохо определены, за исключением случая Chronic Myeloid Leukaemia
(CML), ассоциированной с характерной хромосомной транслокацией 'the Philadelphia chromosome' и ассоциированной с перестроенным геном BCR - ABL.
PV клетки предшественники, как было показано, растут в отсутствие добавления erythropoietin, образование т.наз. endogenous erythroid
colony (EEC) formation1, и гиперчувствительны к различным др. цитокинам, включая insulin-like growth factor-12. Формирование EEC, однако, не специфично для PV и обнаруживается также и при др. MPDs. Др. свойства включают повышенную экспрессию ингибитора апоптоза Bcl-xL
в отсутствие Epo в PV эритроидных клетках, подтверждая, что дерегуляция экспрессии Bcl-xL может вносить вклад в erythropoietin зависимое выживание клеток эритроидного клона при PV.3 Экспрессия thrombopoietin рецептора, Mpl, тромбоцитами и мегакариоцитами у пациентов с PV, как было установлено, редуцируется по сравнению с нормальным контролем.4
Это снова не специфично для PV, однако, и может происходить и при др.
MPDs. Синтез РНК с гена polycythaemia rubra vera 1 (PRV-1) обнаруживает повышенную экспрессию в PV гранулоцитах.5 Эритроидные колонии от PV пациентов, как было показано, содержат гиперактивную ассоциированную с мембранами tyrosine phosphatase PTP-MEG2, хотя её роль в эритрогенезе нуждается в дальнейшем исследовании.6
В нормальных условиях связывание Epo рецепторами своего лиганда индуцирует быстрое фосфорилирование Akt и последующее стимулирование путей для эритроидных колоний. Эритроидные клетки, происходящие из индивидов с PV, как было показано, демонстрируют повышенное фосфорилирование Akt/PKb
а также Glycogen synthase Kinase 3.7 Это вносит вклад в наследуемые свойства жизнеспособности эритроидных клеток. Анализ микромассивов идентифицировал гены кандидаты, вовлекаемые в патофизиологию PV, включая транскрипцию фактора NF-E2 (Nuclear Factor (Erythroid-derived 2)). Было установлено, что он избыточно экспрессируется в костном мозге в мегакариоцитах и миэлоидных и эритроидных предшественниках субъектов с PV
.8
Более 50 лет назад Dameshek
9 связал вместе распознаваемые нарушения PV, chronic myeloid leukaemia и IMF и высказал предположение об общих myelostimulatory факторах. В начале 1970s chronic myeloid leukaemia была отделена как самостоятельное заболевание, определяемое одиночной хромосомной и наконец генной перестройкой (BCR/ABL).
10 Вплоть до недавнего времени др. MPDs продолжали отделяться и диагностироваться на базе соотв. клинических и лабораторных исследований (Table I).
Однако, недавно сделана молекулярная находка гена
JAK2, общего всем этим нарушениям.
JANUS KINASE 2
Ген
JAK2 впервые был клонирован в 1989
11 он является членом семейства из 4-х Janus kinases 1, 2 and 3 и tyrosine kinase 2.
Table I
Diagnostic criteria for common myeloproliferative disorders
Первоначально она была названа 'just another kinase', но белковая группа была переименована в Janus kinases согласно Богу Римлян входа и выхода. Эти не рецепторные киназы имеют два сходных 'active' и 'inactive' домены и это напоминает Бога Януса. который обладает способностью смотреть сразу в двух направлениях.
Каждая JAK имеет активный tyrosine kinase domain, JAK homology 1(JH1), каталитически неактивный псевдокиназный домен, JAK homology 2 (JH2), SRC homology 2 (SH2) домен и N-терминальный homology домен FERM (4-point-1, Erzin, Radixin, Moesin) , где происходит связывание type
1 cytokine рецепторов. Взаимодействия JAK2 FERM домена также выполняют роль по поставке цитоплазматического домена EPO receptor (EPOR) на поверхность клетки.
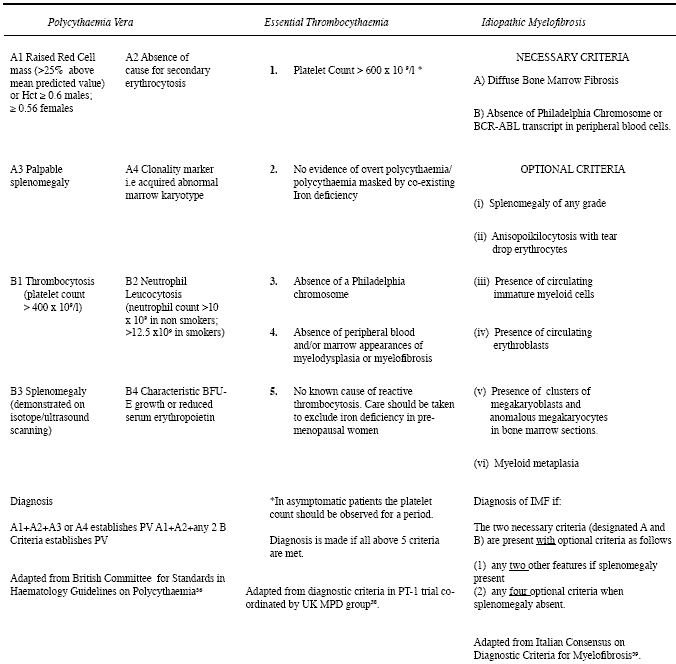
12 При нормальных физиологических условиях, когда лиганд (напр., erythropoietin) связывается с рецептором, то происходит конформационное изменение (fig 1). Белок JAK2 затем устанавливает контакт с цитоплазматическим доменом рецептора, где он катализирует фосфорилирование тирозина. Это в первую очередь ведет к рекрутированию STAT (signal transducer and activator of transcription) молекул, которые затем фосфорилируются, гомодимеризуются и транслоцируются в ядро, где они действуют как транскрипционные факторы. Фосфорилирование тирозина модифицирует также и др. ключевое регуляторное событие, участвующее в путях передачи сигналов цитокинов.
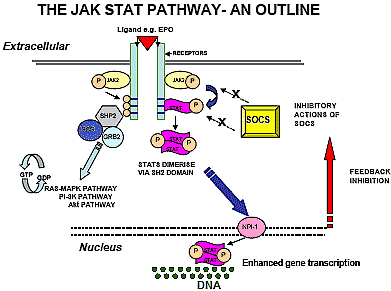 Fig 1. Diagram illustrating Functional JAK STAT Pathway
This "JAK STAT" pathway appears to be ubiquitous
amongst vertebrates. Following ligand binding the
activated JAK2 protein catalyses tyrosine phosphorylation
in the cytoplasmic domain of the receptor and also leads to
phospharylation of the Signal Transducers and Activators
of Transcription (STATS).
Phosphorylation of STAT leads to dimerisation via
conserved Src homology 2 (SH2) domains. Translocation
of these dimers to the nucleus then occurs facilitated via
Nucleoprotein Interactor 1 (NPI .1). Subsequent regulation
of gene expression following interaction with DNA
response elements occurs. This leads to a transcriptional
response. There is also interaction with the RAS/MAPK,
Pl.3 K and Akt downstream pathways.
Under normal conditions the enhanced gene expression is
under complex negative feedback mechanisms including
amongst others the production of the negative regulator
Suppressors of Cytokine Signalling( SOCS).
Fig 1. Diagram illustrating Functional JAK STAT Pathway
This "JAK STAT" pathway appears to be ubiquitous
amongst vertebrates. Following ligand binding the
activated JAK2 protein catalyses tyrosine phosphorylation
in the cytoplasmic domain of the receptor and also leads to
phospharylation of the Signal Transducers and Activators
of Transcription (STATS).
Phosphorylation of STAT leads to dimerisation via
conserved Src homology 2 (SH2) domains. Translocation
of these dimers to the nucleus then occurs facilitated via
Nucleoprotein Interactor 1 (NPI .1). Subsequent regulation
of gene expression following interaction with DNA
response elements occurs. This leads to a transcriptional
response. There is also interaction with the RAS/MAPK,
Pl.3 K and Akt downstream pathways.
Under normal conditions the enhanced gene expression is
under complex negative feedback mechanisms including
amongst others the production of the negative regulator
Suppressors of Cytokine Signalling( SOCS). Домен JH2 является не каталитической псевдокиназой и обладает несколькими критическими регуляторными функциями.13 Очевидно, что в отсутствии связывания лиганда он обладает аутоингибирующими свойствами, большинство из которых проявляется при взаимодействии JH2/JH1 и в случае нарушения этой области происходит дисрегуляция этой аутоингибиции. Очевидно также, что максимальная активность JAK2 в ответ на цитокины нуждается в интактной JH2 области.
В начале 2005 4 разных группы описали идентичные мутации в JAK2 V617F у большого количества пациентов с MPDs.14-17 Хотя все группы добились одного и того же результата, они пользовались разными методами. Группа Vainchenker, который общепризнан как первооткрыватель, использовала подход с точки зрения биологии болезни.14 Перед этим они наблюдали, что ингибиторы JAK2 и др. киназы вмешиваются в независимую от erythropoietin дифференцировку у PV.18 Следовательно, они усмотрели потенциальные механизмы, ведущие к формированию EECs.
Идентифицировав JAK2 в качестве потенциального гена кандидата и поняв ее роль как вышестоящей сигнальной молекулы, непосредственно связанной с erythropoietin рецептором, они сфокусировали внимание на этой тирозин киназе. Они открыли, что подавление регуляторной экспрессии JAK2
путем внесения короткой interfering РНК ведет к заметному ингибированию формирования EEC у индивидов с PV. Это направило исследователей на изучение ключевой роли JAK2 в формировании EEC и помогло секвенировать кодирующие экзоны и интрон - экзонные соединения гена у трех пациентов с PV и у двух нормальных людей. Два индивида с PV продемонстрировали присутствие мутации JAK2 V617F. (fig 2) В дальнейшем большая группа пациентов с PV имела мутацию в 88% случаев. У всех контрольных людей в дополнение ко всем 35 выборкам пациентов со вторичным erythrocytosis, выявлена только дикого типа
JAK2.
 Fig 2. Diagram of JAK2 Domains highlighting main roles and
indicating approximate location of V617F Mutation.
Fig 2. Diagram of JAK2 Domains highlighting main roles and
indicating approximate location of V617F Mutation.
Мутации, как было установлено, являются благоприобретенными, т.к. она присутствует в миэлоидном клоне, но отсутствует в Т клетках. Эта группа идентифицировала также способность мутантной JAK2 спонтанно активировать нижестоящим STAT обусловленную транскрипцию в отсутствии лиганда erythropoietin. Это находится в контрасте с неспособностью дикого типа JAK2 обеспечивать такие события. Обнаруживается также активация ERK/MAP kinase и P13K/AKT путей в отсутствии стимуляции альтернативными цитокинами. Итак, auto-inhibitory активность JAK2 нарушается присутствием этой
V617F мутации.
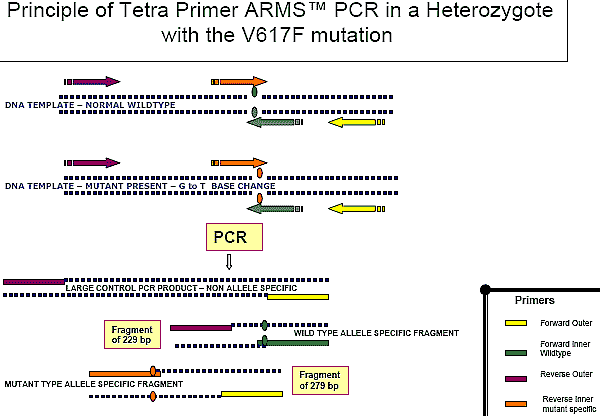 Fig 3. Tetra primer ARMS PCR
o Tetra Primer Amplification Refractory Mutation Screening Polymerase Chain Reaction - ARMS-PCR-
is an extremely efficient
detection method of single nucleotide polymorphisms (SNPS).
o It consists of two pairs of primers - to amplify wildtype and mutant respectively.
o Two amplification allele.specific reactions occur in opposite directions, simultaneously.
o The mismatch is in the middle of the inner primers in contrast to conventional ARMS PCR.
o There is the outer forward primer, the forward inner wildtype primer, the reverse outer primer and the reverse inner mutant
specific primer.
o It requires two temperature programs during the PCR reaction.
o Due to the positioning of the outer primers at varying distance from the site of the mutation there is the generation of three
fragments in this example in a heterozygote: two small allele specific fragments and a large control PCR product.
o DNA Fragments can be distinguished via electrophoresis on agarose gel.
Fig 3. Tetra primer ARMS PCR
o Tetra Primer Amplification Refractory Mutation Screening Polymerase Chain Reaction - ARMS-PCR-
is an extremely efficient
detection method of single nucleotide polymorphisms (SNPS).
o It consists of two pairs of primers - to amplify wildtype and mutant respectively.
o Two amplification allele.specific reactions occur in opposite directions, simultaneously.
o The mismatch is in the middle of the inner primers in contrast to conventional ARMS PCR.
o There is the outer forward primer, the forward inner wildtype primer, the reverse outer primer and the reverse inner mutant
specific primer.
o It requires two temperature programs during the PCR reaction.
o Due to the positioning of the outer primers at varying distance from the site of the mutation there is the generation of three
fragments in this example in a heterozygote: two small allele specific fragments and a large control PCR product.
o DNA Fragments can be distinguished via electrophoresis on agarose gel.
Группа Skoda's
15 отталкивалась от предыдущей работы, они идентифицировали loss of heterozygosity (LOH) в коротком плече хромосомы 9 у части MPD пациентов посредством скрининга микросателлитов по всему геному. Это подтвердило, что 9p м.служить патогенной мутацией. Они первоначально использовали 10 microsatellite маркеров, покрывающих хромосому 9p и выявили 9p LOH в гранулоцитах, происходящих от пациентов с MPDs в 21% (51 /244) случаев и ни одного в контроле, включая CML. Все 51 пациентов с 9p LOH обладали
JAK2 V617F мутацией. Дальнейшие исследования выявили мутацию у 65% пациентов PV из них 27% обладали гомозиготной мутацией и 38% гетерозиготной.
Дисрегуляция ключевых тирозин киназ является первостепенной в патогенезе многих раков, включая CML. Это способствовало дальнейшему углублению поисков мутаций в тирозин киназах при обычных myeloproliferative болезнях. Группа Gilliland's 16 предприняла поиск мутаций тирозин киназ путем использования высокопроизводительного анализа последовательностей и выявила JAK2
V61F мутацию. Как часть большого исследования по анализу генов киназных белков при MPDs группа Green's 17 выявила мутации у
57% индивидов с ET, 50% индивидов с IMF и у 97% с PV. Они были выявлены как в granulocyte macrophage, так и в эритроидных колониях, и что необычно присутствовали во всех EECs, демонстрируя тем самым связь с гиперчувствительностью фактора роста.
Мутация JAK2 V61F объясняет некоторые из аномалий, описанные при PV, хотя молекулярные события, связывающие мутацию с биологическими параметрами нуждаются в дальнейшем уточнении. JH2 домен является псевдокиназой и обладает аутоингибирующими свойствами, которые предупреждают фосфорилирование рецептора. Из модельных исследований высоко законсервированный валин в позиции 617 как предсказывается находится на верхней поверхности N-терминальной доли домена JH2.
Замена валина на крупный фенилаланин дестабилизирует складку домена.
16 Следовательно, присутствие мутации д. приводить к JAK2, которая будет постоянно активной. Интересно, что у гетерозигот, по-видимому, происходит конкуренция между дикого типа и мутантным генами. Гематопоэтические стволовые клетки от MPD пациентов, гиперчувствительных к ряду ростовых факторов, и используют JAK2 для передачи сигналов. Наблюдения подтверждают нарушения передачи сигналов ниже JAK2, включая конституитивную активацию STAT3,
20 позитивную регуляцию анти-апоптического белка Bcl-xL
3 и повышенную активность AKT.
7 Death рецептор, стимулирующий апоптические пути также, по-видимому, нарушен в
JAK2 V617F эритробластах от PV, с разрегулированной экспрессией короткой изоформы c-FLIP
21, которая фундаментально играет существенную роль в нормальном гомеостатическом апоптическом каскаде.
DETECTION METHODS
The JAK2 V617F mutation in MPDs can be detected by a
variety of methods. The simplest method is to isolate DNA
from whole blood leukocytes and use PCR-direct sequencing.
However, since the mutation is acquired and restricted to the
myeloid lineage this method has a sensitivity of between
20 to 30%. By implication the JAK2 V617F mutant clone
would have to constitute a significant proportion of the total
leukocyte population to be detectable by this method.
Ficoll gradient centrifugation can be used to isolate
mononuclear cells, with subsequent separation into
granulocyte/macrophage lineages and lymphocytes. Methods
to ensure an absence of gross contamination of the fractions
should be utilised, for example magnetic sorting/flow
cytometry. Isolation of DNA from the fractions can then
occur and direct sequencing methods instituted. This allows
detection of an acquired JAK2 V617F mutation in cells of
myeloid lineage.
Amplification Refractory Mutation Screening (ARMS) PCR
permits a single base change to be detected under ideal PCR
conditions. This is ideal for detection of the single base G > T
transversion associated with the JAK2 mutation in question.
The ARMS-PCR technique (fig 3) uses 4 primers as follows;
a forward outer primer, a reverse outer primer, a forward inner
wild type specific primer and a reverse inner mutant specific
primer. The forward primer from one set and the reverse from
the other are able to amplify a positive control band. The
other two primers span the site of the JAK2 V617F mutation.
Therefore in the presence of the JAK2 mutation the reverse
inner mutant specific primer and the forward outer primer
bind to give a fragment of 279 bp. In the presence of wild-type
JAK2 the reverse outer primer and the forward inner wild-type
specific primer produce a fragment of 229bp. Performing a
dilution series indicates the level of sensitivity of ARMSPCR
to be 1-2 %.22 The assay allows discrimination between
homozygous and heterozygous individuals with the JAK2
V617F mutation and has a key role in acting as a reliable
screening test for the presence or absence of the mutation in
individuals with MPDs.
Although ARMS-PCR can indicate whether an individual
is homozygous or heterozygous for the V617F JAK2
mutation it does not quantify the ratios of the wild type and
mutant alleles. This also applies to PCR-direct sequencing.
Pyrosequencing which uses a quantitative technique originally
designed for the detection of single nucleotide polymorphisms
(SNP) has been developed for the detection of the G1849T
JAK2 mutation, providing accurate allele ratios.23, 24 This
technique is unique in that it provides a quantitative result for
each allele. Pyrosequencing entails amplification by PCR of
exon 12 of JAK2, with a set of primers where one is labelled
with a biotin tag. Single-stranded PCR products are prepared
post PCR. Any biotin tagged single stranded DNA molecules
are then specifically isolated by capture with streptavidin
beads and genotypes are determined using the SNP software,
which allows allele frequency quantification and scoring of
individuals as homozygous when the mutant allele is greater
than 50%.
CLINICAL CORRELATES
Др. группы также осуществляли наблюдение за своей серией пациентов и выявили сходные доли присутствия мутаций в различных группах больных (Table II).14-17, 22-25 Интересно то, что мутация обнаруживается чрезвычайно редко у тех, у кого не идентифицирована причина erythrocytosis, и скрининг, следовательно, является благоприятным для детекции клональной болезни.26 Присутствие мутации JAK2 V617F коррелирует с др. описанными биологическими феноменами, такими как экспрессия PRV1 и формирование EEC.27 Пациенты с гетерозиготными или гомозиготными мутациями JAK2 V617F, как было показано, экспрессируют высокие уровни NF-E2 по сравнению с индивидами без мутации.28
Те, что гомозиготны по мутации, могут иметь и другие или более прогрессивное заболевание по сравнению с гетерозиготами. Kravolics 15 показал, что те, которые гомозиготны, имели более длительную продолжительность болезни и были более склонны к возникновению вторичного myelofibrosis. Tefferi 29 продемонстрировал, что PV пациенты, обладающие гомозиготной JAK2 мутацией, обнаруживают тенденцию к более высоким уровням гемоглобина, повышенным показателям зуда и более высокой доле fibrotic осложнений.
Важность рассмотрения случаев с очень небольшим процентом
JAK2 мутантных позитивных клонов, как показателем "действительно"
JAK2 V617F позитивных, остается неясной. Очевидно, что необходимо учитывать какие 'cut off ' значения мы используем для отличия тех, что обладают мутациями от тех, что классифицируются как не имеющие мутации. Это важно, если присутствие мутации
JAK2 V61F становится главным для классификации системы MPDs. Семейные MPDs, включая polycythaemia,
30 хорошо документированы и очевидно, что даже в этих редких случаях мутации JAK2, по-видимому, скорее соматические, чем происходящие из зародышевой линии. Значение мутации в IMF, по клиническим данным, по-видимому, несколько хуже определено. В большом исследовании
myelofibrosis, Campbell et al
31 показали, что пациенты с мутацией
JAK2 V617F имеют более высокие количества нейтрофилов и белых кровяных клеток по сравнению с пациентами без мутации и в целом обнаруживают тенденцию к более плохому прогнозу. Tefferi et al
32 занимались поиском мутации у разных пациентов с myelofibrosis и нашли, что мутация скорее всего присутствует у пациентов с PV в анамнезе по сравнению с
de novo MF. Присутствие мутации
JAK2 V617F в этой довольно крупной кагорте из 157 пациентов, по-видимому, не имеет прогностического значения.
Table II
Some reported incidences of JAK2 V61F mutation in myeloproliferative disorders
 KEY TO ABBREVIATIONS
PV: polycythemia vera, ET: essential thrombocythemia, IMF: idiopathic myelofibrosis, SM: systmemic mastocytosis, CNL: chronic neutrophilic
leukemia, HES: hypereosinophilic syndrome, UN: unclassified MPD, MDS: myelodysplastic syndrome, CMML: chronic myelomonocytic
leukemia.
KEY TO ABBREVIATIONS
PV: polycythemia vera, ET: essential thrombocythemia, IMF: idiopathic myelofibrosis, SM: systmemic mastocytosis, CNL: chronic neutrophilic
leukemia, HES: hypereosinophilic syndrome, UN: unclassified MPD, MDS: myelodysplastic syndrome, CMML: chronic myelomonocytic
leukemia.
MRC-PT-1 проспективное исследование ET позволило сравнить позитивных и негативных по мутации пациентов.
33 Те, что с мутацией, имели признаки, напоминающие PV, высокий гемоглобин, высокое количество нейтрофилов, множество тромбозов вен и высокая доля трансформации в PV, но они имели более низкие уровни сывороточного erythropoietin и ferritin.
JAK2 V617F позитивные пациенты, по-видимому, более чувствительны к лечению hydroxycarbamide, но не anagrelide. Др. серия ET пациентов показала, что те, что с мутацией, более легко трансформировались в PV.
34 Эти находки ставят вопрос о разделении болезней PV и ET. Может существовать континуум болезни с эффектами
JAK2 V617F мутации на клиническое проявление, зависящее от влияния др. модификаторов, включая потребление железа, подавление erythropoietin и др. генетических модификаторов (см. fig 4)
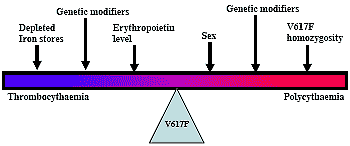 Fig 4. Continuum model of JAK2 V61F Disease.
Fig 4. Continuum model of JAK2 V61F Disease.
OTHER DISEASES
Присутствие мутации провелялось при атипических MPDs, включая systemic mastocytosis, hypereosinophilic syndrome, chronic neutrophilic leukaemia, unclassified myeloproliferative disorders.
24 McLornan et al
35 выявили мутацию у одного пациента с chronic neutrophilic leukaemia с необычным затяжным течением, по-видимому, в некоторых случаях мутация может влиять на течение болезни. Мутация была найдена в варьирующих пропорциях у пациентов, суммированных в Table II. Она обнаруживается редко у пациентов с myelodysplastic disorders.
22 Присутсвие мутации высоко у пациентов с acute myeloid leukaemia (AML) с предшествующей PV или IMF, чем в общей кагорте.
36 Она описывается редко в др. сериях пациентов с AML и отсутствует у пациентов с lymphoid leukaemia.
23, 36
FURTHER QUESTIONS
The fascinating discovery of a single mutation in a wide
spectrum of MPDs has lead to rapid progress in the
investigation of MPDs but leads to a number of further
questions. The hierarchical position of this mutation requires
further study. It is still not clear whether the JAK2 V61F
mutation is the primary initiating event or a secondary event
with an as yet unknown primary event. While the presence of
the mutation was sufficient to induce erythrocytosis in mice
this is a manipulated experimental situation and it may not be
sufficient to initiate disease in the human.
Classification of MPD needs revision. The presence of
the mutation demonstrates a clonal disorder but splitting
of diseases on the basis of clinical characteristics needs reconsideration. Questions remain about the
underlying pathogenesis in those with mutation negative
myeloproliferative disorders. In summary, the discovery of a
single mutation JAK2 V61F in a large number of MPD patients
has lead to great progress in the understanding of MPDs but
leads to many more exciting biological questions.
Сайт создан в системе
uCoz