Посещений: 

АУТИЗМ
Генетический Контроль
|
|
Insights into the Pathogenesis of AutismJames S. Sutcliffe  SCIENCE, VOL 321, P.208-209, 2008. www.sciencemag.org
|
Genetic analysis of inbred families reveals genes associated with susceptibility to autism.
Identifying Autism Loci and Genes by Tracing Recent Shared Ancestry
Eric M. Morrow, Yingxi Lin, Seung-Yun Yoo, Robert Sean Hill, Generoso Gascon,
Robert M. Joseph, Kira A. Apse, Asif Hashmi, Steven W. Flavell, Nahit M. Mukaddes, Samira Al-Saad, Rachel Greenblatt, Adria Bodell, Kyriacos Markianos,
Tae-Kyung Kim, Soher Balkhy, Janice Ware, Danielle Gleason, Jennifer N. Partlow, Russell J. Ferland, Julia A. Ertelt, Brenda Barry, Michael E. Greenberg, Hui Yao, Christopher A. Walsh Science, V. 321, P.218-223, 2008
To find inherited causes of autism-spectrum disorders, we studied families in which parents share ancestors, enhancing the role of inherited factors. We mapped several loci, some containing large, inheri te d, homozygous de le ti ons that a re likely muta ti ons. The l ar ge st deletions i mp li ca te d ge ne s, inclu ding PCDH10 ( protocadherin 10)and DIA1 (deleted in autism1 ,or c3orf58),whose level of expression changes in response to neuronal activity, a marker of genes involved in synaptic changes that underlie learning. A subset of genes, including NHE9 (Na +/H + exchanger 9 ), showed additional potential mutations in patients with unrelated parents. Our findings highlight the utility of “ homozygosity mapping ” in heterogeneous disorders like autism but also suggest that defective regulation of gene expression after neural activity may be a mechanism common to seemingly diverse autism mutations.
|

Autism Spectrum Disorders |
| |

Аутизм широко распространенное заболевание, которое существенно нарушает социальное поведение и общение детей до 3-летнего возраста. Повторяющееся, стереотипное и навязчиво-компульсивное поведение является выделяющимся признаком нарушения (1) оно часто сопровождается нарушением познавательной функции, судорогами или эпилепсией, желудочно-кишечными жалобами, нарушениями сна и др. проблемами. Идентификация факторов риска аутизма является приоритетной задачей для ученых, lay groups и родителей аутентичных детей. Morrow et al. (2) добавили еще несколько генов ко все увеличивающемуся количеству генетических аномалий, коррелирующих с чувствительностью к аутизму. Исследования близнецов и семей показали, что этиология аутизма имеет существенный генетический компонент. Современные подсчеты риска рецидивов у братьев сестер показали, что вероятность у молодых братьев, сестер ребенка с аутизмом также иметь аутизм выше 15%
Сравнение с популяционным показателем примерно1 на 500 детей для узко определяемого аутизма, 1 на 150 детей для более широко определяемого спектра аутических нарушений указывает на высокую степень наследуемости в семьях. Определение специфических генетических изменений, которые увеличивают риск развития нарушений, подобных аутизму чрезвычайно сложно (6) из-за гетерогенности - вовлечены разные типы изменчивости многих лежащих в основе генов. Один тип изменчивости состоит из редкой болезнетворности или высоко пенетрантных мутаций и это влияет на специфические биологические процессы. Сходным образом, общая изменчивость - обычно дискретных изменений в последовательностях ДНК - идентифицирована при аутизме, но воспроизведены лишь немногие специфические находки. Др. важным показателем генетических факторов при аутизме являются аномалии, такие как хромосомные транслокации, инверсии и крупные делеции или дупликации, которые более частые у индивидов, которые характеризуются клинически дизморфными признаками и тяжелым нарушением познавательной способности. Генетики уже давно предполагали, что гены, нарушаемые хромосомными аномалиями в отдельных случаях могут играть более широкую роль в
PUTATIVE AND KNOWN AUTISM-RELATED GENES
Glutamatergic synapse function and/or
neuronal cell adhesion
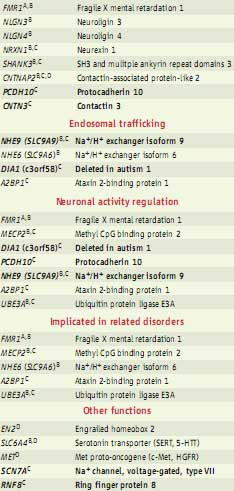 Genes implicated in autism pathogenesis. Genes have been implicated in autism (1, 2) on the basis of different functions and forms of genetic variation, and also on their association with disorders that show features of autism. They share common or related pathways, as shown. A, genes showing triplet repeat expansion; B, genes with rare mutations or coding variants; C, genes with copy number variation or chro- mosomal abnormality; D, association of common alleles. Genes implicated from (2) are shown in bold. Genes implicated in autism pathogenesis. Genes have been implicated in autism (1, 2) on the basis of different functions and forms of genetic variation, and also on their association with disorders that show features of autism. They share common or related pathways, as shown. A, genes showing triplet repeat expansion; B, genes with rare mutations or coding variants; C, genes with copy number variation or chro- mosomal abnormality; D, association of common alleles. Genes implicated from (2) are shown in bold.
восприимчивости к аутизму. Недавние успехи технологии микромассивов ДНК выявили существенную этиологическую роль малых потерь и добавок ДНК - т.наз вариации в количестве копий - при аутизме (7-12). Все индивиды обладают этой распространенной формой генетической изменчивости, которая может наследоваться от родителей или может возникать спорадически de novo. Однако, огромное и всё увеличивающееся количество делеций и дупликаций ДНК было обнаружено у людей с аутизмом. По сравнению с контрольными выборками необходимо идентифицировать, какие варианты являются уникальными, более частыми или равными при аутизме и в контроле. тогда нам будет легче интерпретировать наблюдаемые вариации в числе копий. Большинство споров концентрируется вокруг того, наследуется ли вариант числа копий или возникает de novo, с более высоким интерпретируемым весом vis-а-vis болезненных ассоциаций, присущих последнему. При крупных хромосомных аномалиях возможно, что нарушение или дерегуляция экспрессии генов лежит в основе риска или вызываемого эффекта для данного варианта количества копий. Гены могут быть потеряны или присутствовать в дополнительной копии на данной хромосоме; гены, фланкирующие делеции или дупликации ДНК может быть предметом нарушения регуляции из-за измененной локально структуры хроматина или из-за отделения от ключевых энхансерных элементов (которые регулируют генную экспрессию). Т.о.. изменчивость в числе копий является главной категорией генетического риска спектра аутических нарушений и проявляет себя в 10-20% (или более) случаев Генетическая гетерогенность аутизма существенно осложняет идентификацию генов, которые повышают чувствительность к этой болезни. Morrow et al. использовали мощную генетическую технику homozygosity mapping для идентификации генов аутизма. Генетики уже дано используют преимущества силы статистики, дающей возможность генетического анализа семей, в которых родители затронутых индивидов обнаруживают общее происхождение (напр.. двоюродные родственники). Такие генетически родственные семьи более широко встречаются на Среднем Востоке, у них существенно повышен риск аутосомно рецессивных заболеваний [признаки проявляются, когда индивид гомозиготен (имеет две идентичные копии) по определенному гену]. Растет понимание, что инбредные семьи также пригодны для идентификации генов для сложных нарушений, таких как аутизм. Morrow et al. использовали микромассивы ДНК для изучения многочисленных кровнородственных семей со Среднего Востока. Путем анализа наследования ДНК всего генома у таких родословных, они идентифицировали хромосомные регионы, которые наследуются часто затронутыми индивидами, которые обладают теми же самыми двумя копиями этих регионов. Такие гомозиготные сегменты, которые гетерозиготны у соотв. родителей, скорее всего и представляют собой причинный фактор или фактор риска. В некоторых из таких семей регионы сцеплены со спектром аутических нарушений и наследуют "identical by descent" , содержащие делеции. Т.о., затронутые индивиды оказывались полностью дефицитными по генам (или потенциальным регуляторным ДНК), которые расположены внутри делетированных интервалов. В широком смысле отсутствие их генных продуктов и/или возможно измененная экспрессия генов в непосредственной близи от делеции, как предполагается и вызывают спектр аутических нарушений в данной семье. Важным вопросом является, может ли ген, идентифицированный как причина болезни в одиночной инбредной семье, иметь какое-либо отношение к аутизму в неродственных семьях. Кроме того, установление какой ген (или гены) лежит внутри или вблизи делетированного интервала - нарушение которого вызывает болезнь - не является тривиальным. Напр., история одного из таких регионов хромосомы 3q, содержащей крупную (~886 kilobase) делецию. Ген наз. DIA1 (deleted in autism1; известный также как c3orf58) кодирует не охарактеризованный белок, был полностью удален, в то время как NHE9 (Na +/H + exchanger 9), соседний ген, кодирующий мембранный белок, который обменивает внутриклеточный H ++ на внеклеточный Na, остается интактным, но его регуляция нарушена. Чтобы оценить отношение этого гена к аутизму Morrow et al. секвенировали кодирующие области NHE9 у затронутых субъектов от неродственных U.S. семей и обнаружили мутацию потери функции в одной семье. Сходные мутации вызывают эпилептический фенотип у мышей, а мутация родственного NHE6 гена вызывает фенотип с аутичными симптомами и эпилепсией. Кроме того, и др. вариации оказывают влияние, т.к. концентрация внимания на семьях с эпилепсией привела авт. к наблюдению значительно большего количества кодирующих вариантов в случаях по сравнению с контролем. Все эти находки подтверждают, что нарушение регуляции NHE9 является оказывающим вклад или причинным фактором в этой семье. Наиболее интересные наблюдения этого исследования подчеркивают важность функционального класса генов, участвующих в восприимчивости к аутизму. Авт. показали, что некоторые гены, идентифицированные в или скорее затрагиваемые гомозиготными делециями регулируются с помощью нейрональной активности - т.е. их экспрессия изменяется в ответ на стимуляцию активности нейронов. Т.к. аутизм является нарушением нейрального развития, то акцент необходимо сделать на развитии родителей, которое управляется с помощью врожденных паттернов генной экспрессии. Головной мозг продолжает развиваться в течение длительного времени после рождения, однако, опыт и влияние среды играют важную роль в последующем развитии. Синапсы (соединения между нейронами) частично созревают как функция зависимой от опыта нейрональной активности и сопровождаемой её изменений генной экспрессии. Но если гены нарушены мутациями или изменением числа копий, то следует ожидать, что процесс развития синапсов, регулируемый активностью, сам по себе будет нарушен каким-то образом. В самом деле, это гипотеза авторов. Дерегуляция синаптического развития является установленной идеей исследованиями аутизма. Хотя концептуально это огромный шаг, но авт. осторожны в своих заключениях, возможно, что дерегуляция этих генов вызывает нарушения развития синапсов в ответ на влияние среды и опыта в раннем периоде жизни, но это интригующее предположение требует экспериментальных доказательств.
Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmissionSung Han, Chao Tai, Ruth E. Westenbroek, Frank H. Yu, Christine S. Cheah, Gregory B. Potter, John L. Rubenstein, Todd Scheuer, Horacio O. de la Iglesia & William A. Catterall Nature 489 (7416), 385–390 (20 September 2012) doi:10.1038/nature11356
Hyperactivity, anxiety and stereotypies in Scn1a+/- mice
Scn1a+/- мыши обнаруживают дефицит социального взаимодействия. Scn1a+/- мыши обнаруживают дефицит в контекст-зависимой пространсвенной памяти. Условно мутантные Scn1a+/- мыши обладают содным с аутизмом поведением. Дефицит NaV1.1 каналов нарушает трансмиссию GABAергичных нейронов. Лечение родственного аутизму фенотипа Scn1a+/- мышей clonazepam
Гаплонедостаточность по гену SCN1A, кодирующему напряжением управляемому натриевый канал NaV1.1 вызывает синдром Dravet’s, детское нейропсихиатрическое заболевание, включающее повторяющиеся, неподдатливые судороги, дефицит когнитивной функции и поведение спектра аутизма. Нейральные механизмы, ответственные за когнитивный дефицит и поведение спектра аутизма при синдроме Dravet’s, изучены плохо. Здесь мы описываем мышей с гаплонедостаточностью по Scn1a, вызывающей гиперактивность, стереотипированное поведение, дефицит социального общения и нарушенную контекст-зависимую пространственную память. Обонятельная чувствительность сохраняется, но запах новой пищи и социальные ароматы вызывают отвращение у Scn1a+/- мышей. GABAergic нейротрансмиссия специфически нарушена при этой мутации, а селективная делеция NaV1.1 каналов в промежуточных нейронах переднего мозга достаточна, чтобы вызывать эти поведенческие и когнитивные нарушения. Удивительно, лечение низкими дозами clonazepam, позитивным аллостерическим модулятором рецепторов GABAA, полностью устраняет аномальное социальное поведение и дефицит бестрашия у мышей, моделирующих синдром Dravet’s, демонстрируя, что он вызывается за счет нарушения GABAergic нейротрансмиссии, а не повреждения нейронов от повторяющихся судорог. Эти результаты демонстрируют критическую роль NaV1.1 каналов в нейропсихиатрических функциях и открывают потенциальную терапевтическую стратегию для когнитивного дефицита и поведения спектра аутизма при синдроме Dravet’s.
Рисунки к статье
|
Сайт создан в системе
uCoz)