Congenital heart disease (CHD) затрагивают 1-2% от всех детей и являются главной причиной смерти детей до 1 года(1). Среди затронутых детей, по крайней мер,10% нуждаются в хирургическом вмешательстве во время младенчества или детства. Сегодня хорошо известно, что некоторые заболевания сердца с началом у взрослых, такие как болезни аортальных клапанов, обусловленные bicuspid aortic valve (BAV), имеют свои корни в детстве. Как можно объяснить столь высокий показатель? Многочисленные эпидемиологические исследования подчеркивают главную роль генетических и эпигенетических факторов в патогенезе этих заболеваний (2). В последнюю декаду достигнут огромный прогресс в выяснении молекулярных событий, управляющих кардиогенезом, В самом деле, эксперименты на некоторых модельных системах, включая мышей, лягушек и рыбок данио, привели к идентификации многочисленных генов, необходимых для нормального развития сердца и помогли установить иерархию генетических программ для разных морфогенетических событий.
В противовес этим впечатляющим успеха в основных науках, перевод этих знаний в клинические инструменты всё ещё находятся в ранней фазе. Недавнее единодушное мнение можно рассматривать как первую ступень в направлении подходов, которые благоприятствуют генетическим по сравнению с морфологическими алгоритмами (3). Исчерпывающая катологизация всех новых кардиальных генов, идентифицированных у животных моделей или всех новых генотип-фенотипических корреляций у людей не является основой данного обзора. Вместо этого я сконцентрируюсь на избранных вопросах, которые лучше всего представляют возникающие парадигмы и подходы, наиболее влияющие на клиническую кардиологию с точки зрения биологии развития.
Cardiac morphogenesis and its relation to CHD
Формирование сердца является сложным процессом, который высоко консервативен у позвоночных. У эмбрионов человека первичная полоска развивается на 13-й день, а гаструляция происходит на 16-й день. Серп мезодермальной ткани сливается по первичной полоске, чтобы сформировать первичную сердечную трубку на 20-й день. В то же самое время формируется сосудистая сеть в эмбриональном диске и сердце начинает сокращаться на 21-23 день. Петлеобразование начинается на 23 и завершается на 28-й день. В это время возникает межжелудочковая перегородка в виде небольшого гребня на дне общего желудочка, оба желудочка начинают расти и появляются эндокардиальные подушки. На 29-й день образуются truncal swellings (вздутия). На 32/33 день атриовентрикулярный канал сужается и формируется ostium secundum с помощью свободного края первичной перегородки. В конце 7-ой недели развития формируется межжелудочковая перегородка и тракт оттока оказывается полностью разделенным.
Такая последовательность событий в точности воспроизводится у эмбрионов мышей и кур. У этих моделей развитие сердца традиционно описывается как сегментная модель, согласно которой небольшие единицы клеток вдоль первичной полоски образуют пре-паттерн. Индивидуальные сегменты линейной сердечной трубки позднее превращаются в разные части зрелого сердца, при этом передние части первичной полоски формируют артериальный полюс, а задняя часть формирует венозный полюс сердца (4).
В подкрепление этой концепции более ранние исследования уже упоминали о вкладе клеток извне примитивной полоски в будущее сердце (5-7). Современные генетические инструменты, в частности, исследования репортерных генов под контролем промоторов Fgf1O, Mef2c и Isl1 , позволили в точности проследить вклад из дополнительных источников миокардиальных клеток в развивающееся сердце (8-12).
Эти исследования и предоставили сегодняшнюю модель развития сердца. согласно которой разные группы клеток миокардиальных предшественников - в частности, first heart field (FHF) и second heart field (SHF) - вносят вклад во взрослое сердце высоко регулируемым пространственно способом (Fig. 1). Клетки FHF формируют собственно кардиальный серп, превращаясь в линейную сердечную трубку и исключительно формируют левый желудочек (13, 14). Клетки SHF являются недифференцированными миокардиальными предшественниками, которые локализуются в фарингеальной мезодерме, медиальнее и каудальнее сердечной трубки (Fig. 1). Позднее они мигрируют в переднюю часть сердечной трубки и вносят вклад в тракт оттока, правый желудочек и в область притока, как было показано с помощью клонального анализа (13, 15). Эти элегантные исследования предоставили доказательства. что предшественники из FHF и SHF преимущественно занимают области, описанные выше и что оба клона сегрегируют из общей популяции предшественников до стадии кардиального серпа (13). Сравнительно недавно открыто третье поле на венозном полюсе сердца (16). На стадии сердечного полумесяца это поле сердца располагается вентральнее развивающейся сердечной трубки; позднее оно дает рога венозного синуса и периферию тракта притока. На молекулярном уровне это поле строго экспрессирует
Tbx18, но лишены экспрессии
Isl1 и Nkx2.5 , ключевых транскрипционных факторов, необходимых для развития сердца. Мыши Tbx18
-/- не способны формировать рога синуса из периферической мезенхимы и имеют дефектные полые вены, тогда как атриальные структуры нормальны. Помимо молекулярных доказательств каудального поля сердца, это исследование также разрешило вопрос, могут ли кардиальные клетки оказаться детерминированными в миогенные клоны, используя разные пути в зависимости от их пространственного распределения в сердце.
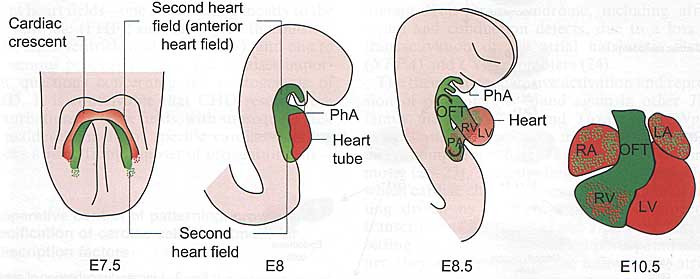 |
Fig. 1. Contribution of two heart fields to the developing heart. The contribution of the second heart field is shown in green, with the anterior heart field subdomain in dark green, and is compared with the myocardial cells that are derived from the first heart field (shown in red). Frontal views are shown for embryonic day 7.5 (E7.5) and E10.5 and lateral views for stages E8 and E8.5. LA, left atria; LV, left ventricle; OFT, outflow tract (D, distal; P, proximal); PA, primitive atria; PhA, pharyngeal arches; RA, right atrium; RV, right ventricle. Reprinted from (105).
|
Fig. 1. Contribution of two heart fields to the developing heart. The contribution of the second heart field is shown in green, with the anterior heart field subdomain in dark green, and is compared with the myocardial cells that are derived from the first heart field (shown in red). Frontal views are shown for embryonic day 7.5 (E7.5) and E10.5 and lateral views for stages E8 and E8.5. LA, left atria; LV, left ventricle; OFT, outflow tract (D, distal; P, proximal); PA, primitive atria; PhA, pharyngeal arches; RA, right atrium; RV, right ventricle. Reprinted from (105).
Среди множества различных маркеров, специфически предназначенных для FHF или SHF, имеется ли единственный маркер, чья экспрессия д. предшествовать всем др. маркерам? Др. словами, существует ли одиночная плюрипотентная стволовая клетка для сердца? Кардиальные стволовые клетки д. быть способны дифференцироваться в такие разные фенотипы, как эндотелиальные, гладкомышечные клетки, клетки проводящей системы и кардиомиоциты. Несколько значимых исследований опубликовано в последние годы, показавших, что кардиальные стволовые клетки являются мультипотентными, Isl1-позитивными мезодермальными предшественниками (17-19) (Fig. 2). Они соответствуют необходимым условиям, быть клональными, само-обновляться и дифференцироваться во множественные различные кардиальные клоны. В частности, они способны генерировать как FHF, так и SHF клетки, исходя из взаимно исключающих паттернов экспрессии маркеров для FHF (Tbx5) и SHF (Isl1) (17) и в соответствии с исследованиями по ретроспективному клональному мечению (13). Конечно, Isl1-позитивные клетки всё ещё могут обнаруживаться в постнатальном и взрослом сердце, совместимые с пулом присутствующих клеток предшественников, участвующих в гомеостазе сердца (19).
Др. уровень сложности добавляется к модели полевой организации и развития сердца недавним более детальным анализом мутантного фенотипа Smad1 и Nkx2.5-дефицитных мышей. В то время как Nkx2.5-нулевые мыши обнаруживают укороченный тракт оттока, гипоморфные Nkx2.5 аллели развиваются далее и обнаруживают уродства, сходные с таковыми при CHD людей, такими как дефекты желудочковой перегородки и двойной вход в правый желудочек. Интересно, что молекулярными признаками этих мутантов была ранняя избыточная экспрессия некоторых генов кардиальных предшественников, таких как Isl1, и секретируемых факторов, таких как Fgf10 и Bmp2. Это указывает на то, что Nkx2.5 регулирует баланс кардиальных предшественников в соотв. полях сердца, путем ограничения спецификации кардиомиоцитов и стимуляции пролиферации SHF посредством передачи сигналов Bmp2/Smad1. Соотв., делеция аллеля Smad1 у Nkx2.5 мутантных мышей ведет к дозово-зависимому устранению морфологических фенотипов. При экстраполяции к известным эффектам модификаторов у мышиных и человеческих моделей CHD, этот пример представляет одиночный каскад вместе с эпистазом по множественным аллелям, что может объяснить варьирующую пенетрантность и экспрессивность у людей с CHD.
Как может примирить модели клонального анализа, линейной сердечной трубки и образования раздутых камер? Ранние описания морфологических процессов развивающегося сердца человека более следовали модели вздутия (ballooning), чем сегментации [Fig. 3 и (20)]. Расхождение между моделями линейной сердечной трубки и полей сердца лучше всего разрешается путем наблюдения за развитием камер, как процессом регионализованной спецификации и пролиферации (21). Эта модель согласуется также с доказательствами, полученными на эмбриональных сердцах кур, которые обнаруживают хорошо организованную пространственно-временную последовательность пролиферации, дифференцировки и увеличения клеточных размеров региональных клеток. Не удивительно, что самые крупные клетки обнаруживаются на вентральной стороне сердечной трубки, месте образования желудочка, демонстрируя тем самым, что региональное увеличение
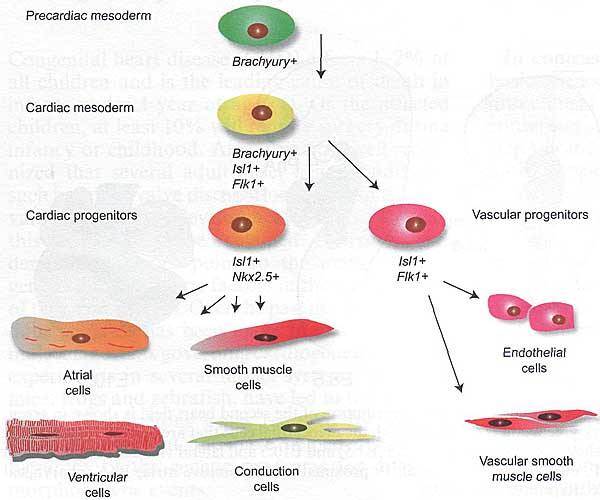 |
Fig. 2. Lineage diversification of cardiac progenitors. From cardiac mesoderm arise multipotent cardiac progenitor cells that give rise to subpopulations with specific molecular signatures and phenotypes. Adapted from (106).
|
Fig. 2. Lineage diversification of cardiac progenitors. From cardiac mesoderm arise multipotent cardiac progenitor cells that give rise to subpopulations with specific molecular signatures and phenotypes. Adapted from (106).
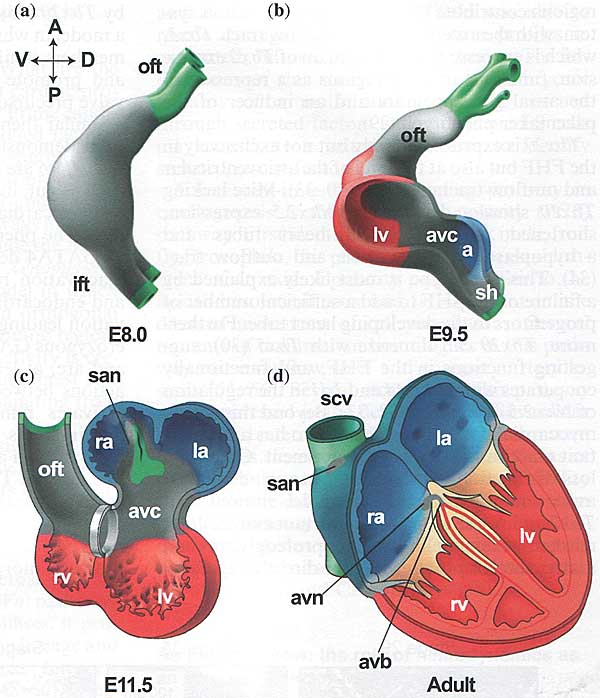 |
Fig. 3. Ballooning model of heart development. Chamber myocardium (red, ventricular; blue, atrial) balloons at the outer curvatures, but not at the inner curvatures of the primary heart tube. Non-chamber myocardium is depicted in gray and does not balloon. The top panels show a left lateral view. Sinus horn myocardium gives rise to the sinoatrial node (san), atrioventricular canal myocardium to the atrioventricular node (avn) and atrioventricular junction. Abbreviations: A, anterior; D, dorsal; P, posterior; V, ventral; a, atrium; avb, atrioventricular bundle; avc, atrioventricular canal; avn, atrioventricular node; ift, inflow tract; sh, sinus horns; oft, outflow tract; la, left atria; lv, left ventricle; ra, right atrium; rv, right ventricle. E = embryonic day. Reprinted from (107).
|
Fig. 3. Ballooning model of heart development. Chamber myocardium (red, ventricular; blue, atrial) balloons at the outer curvatures, but not at the inner curvatures of the primary heart tube. Non-chamber myocardium is depicted in gray and does not balloon. The top panels show a left lateral view. Sinus horn myocardium gives rise to the sinoatrial node (san), atrioventricular canal myocardium to the atrioventricular node (avn) and atrioventricular junction. Abbreviations: A, anterior; D, dorsal; P, posterior; V, ventral; a, atrium; avb, atrioventricular bundle; avc, atrioventricular canal; avn, atrioventricular node; ift, inflow tract; sh, sinus horns; oft, outflow tract; la, left atria; lv, left ventricle; ra, right atrium; rv, right ventricle. E = embryonic day. Reprinted from (107).
размеров миоцитов вносит существенный вклад в формирование камер (22). Взятое всё вместе, идентификация трех разных полей сердца - одно вносит наибольший вклад в левый желудочек (FHF); одно в основном в тракт оттока, правый желудочек и предсердия (SHF); и одно в венозный полюс (Tbx18 позитивный) - ставит важные вопросы, связанные с патогенезом CHD. Вполне возможно, что CHD возникают в результате пертурбаций в этих полях, с последующим отсутствием или неправильным развитием специфических кардиальных строительных блоков, возникающих из субнаборов клеток предшественников.
Cooperative control of patterning, growth and specification of cardiac cells: a leitmotif for transcription factors
Морфогенез сердца нуждается в исполнении сложной генетической программы, в которой транскрипционные факторы оккупируют ключевые позиции. Многочисленные транскрипицонные факторы кооперируются, чтобы контролировать формирование паттерна сердца. Одним из первых транскрипционных факторов, идентифицированных для контроля за этим процессом стал Tbx5, который мутантен в 75% случаев синдрома Holt-Oram (23). Мыши, лишенные одного из аллелей Tbx5 воспроизводят кардиальные признаки, свойственные синдрому Holt-Oram, включая дефекты межпредсердной перегородки и проводящей системы, обусловленные потерей трансактивациии промоторов atrial natriuretic factor (NPPA) and Cx40 (24).
Вопрос о кооперативной активации и репрессии промоторов возникает снова в отношении др. членов семейства Tbx: Tbx2 и Tbx3 репрессируют экспрессию Nppa в первичном миокарде за счет непосредственной конкуренции с Tbx5 за промотор Nppa (25-27). Эти находки подтверждают модель, согласно которой образование сердечных камер является используемым по умолчанию процессом, управляемым с помощью строго экспрессируемых кардиогенных транскрипционных факторов. Tbx2 и Tbx3 устраняют эту установку четко регулируемым пространственно-временным способом: они экспрессируются в тракте притока, атриовентрикулярном канале, внутренней кривизне и тракте оттока, где они ингибируют экспрессию Cx40 и Nppa, необходимых для дифференцировки и Nmycl и cyclin A2 (Ccnal), необходимых для пролиферации (28). Интересно отметить, что все эти регионы вносят вклад в проводящую систему сердца, за исключением тракта оттока. Tbx3, который экспрессируется в субдомене экспрессии Tbx2 , действует в этих регионах как репрессор атриальных генетических программ и как индуктор фенотипа синусового узла (29).
Tbx20 экспрессируется главным образом, но не исключительно в FHF, но также на уровне атриовентрикулярных подушках и подушках тракта оттока (30-33). Мыши, лишенные Tbx20 обнаруживают снижение экспрессии Nkx2.5, укороченную, недоразвитую сердечную трубку и гипопластический правый желудочек и тракт оттока (34). Этот фенотип лучше всего объясняется неспособностью SHF добавлять достаточные количества предшественников для развития сердечной трубки. Более того, Tbx20 can может димеризоваться с Tbx5 (30), подтвержая функцию в FHF, и функционально кооперирует с GATA4 и Isl1 в регуляции экспрессии Nkx2.5 (32, 33). Помимо этой роли в формировании миокардиального паттерна, Tbx20 также играет главную роль в развитии сердечных клапанов. Эксперименты с моделью избыточной и недостаточной функции в эксплантированных эндокардиальных подушках показали, что Tbx2 и Nmyc также как и важные внеклеточные маркеры ремоделирования, такие как proteoglycans и матричные metalloproteinases, непосредственно регулируются с помощью Tbx20. Это согласуется с моделью, согласно которой взаимодействия с членами семейства Tbx ингибируют ремоделирование и способствуют клеточной пролиферации в популяциях мезенхимных предшественников клапанов.
Сходная тема кооперативного взаимодействия была продемонстрирована для членов семейства GATA. Gata4/5/6 экспрессируются в мезодермальных тканях, таких как кишечник, печень легкие и сердце.
Gata4-нулевые мыши обнаруживают cardia bifida и являются эмбриональными леталями (36). Фенотипы кардио-специфческой делеции GATA4 зависят от временной точки, при ранней инактивации возникает миокардиальная гипоплазия и дефекты эндокардиальных подушек, а при поздней инактивации наблюдается снижение функции (37, 38). Гетерозиготные GATA4 мутации вызывают у людей CHD и объясняются, по крайней мере, частично за счет взаимодействий между TBX5 и GATA4 (39). Gata4 активирует многочисленные кардио-специфические промоторы и взаимодействует с многочисленными др. транскрипционными факторами, такими как Nkx2.5, Tbx5, Isl1 и Kriippel-like факторами. Эти примеры иллюстрируют, как кооперативные взаимодействия между транскрипционными факторами необходимы в течение всего развития сердца и как пертурбации этих процессов вызывают драматические морфологические последствия (Fig. 4).
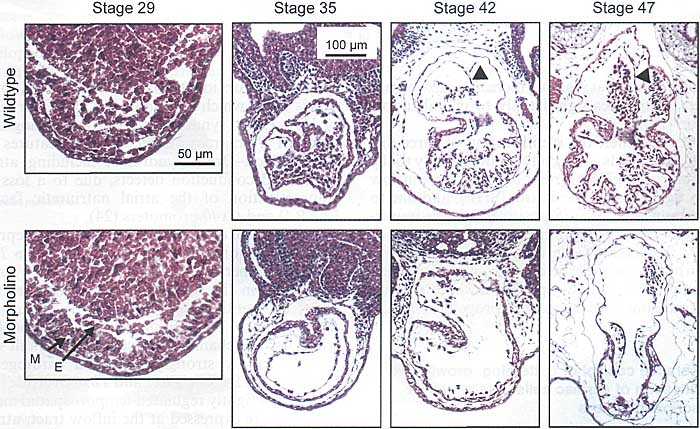 |
Fig. 4. Morphological consequences of knockdown of KLF13 in Xenopus embryos. Top row: Normal development of the Xenopus heart (Stages 29-47). Bottom row: Heart development is perturbed upon loss of KLF13 in Xenopus, with disorganized linear heart tube (Stage 29), normal looping (Stage 35), absence of trabecularization of the ventricle (Stage 42), absence of interatrial septum and thin, small ventricle (Stage 47). Arrows: M, myocardium; E, endocardium. Arrowheads: Interatrial septum.
|
Fig. 4. Morphological consequences of knockdown of KLF13 in Xenopus embryos. Top row: Normal development of the Xenopus heart (Stages 29-47). Bottom row: Heart development is perturbed upon loss of KLF13 in Xenopus, with disorganized linear heart tube (Stage 29), normal looping (Stage 35), absence of trabecularization of the ventricle (Stage 42), absence of interatrial septum and thin, small ventricle (Stage 47). Arrows: M, myocardium; E, endocardium. Arrowheads: Interatrial septum.
Genetics and congenital heart disease
Signaling events in cardiogenesis: timing is everything
Как же начинается образование сердца? Какие сигналы запускают экспрессию маркеров кардиальных предшественников и на какой самой ранней стадии? первыми кандидатами для такой регуляции являются секретируемые факторы, которые д. пересекаться в виде разных временных и пространственных паттернов, чтобы специфицировать детерминацию миокардиальных предшественников до начала экспрессии Isl1 и Nkx2.5. Главными сигнальными путями, участвующими у позвоночных в азвитии сердца являются BMP, TGF-β , FGF и Wnt пути. Xenopus является удобным организмом для описания модели, в которой эти сигнальные факторы перекрываются ортогональным способом в презумптивной сердце-образующей области: так что область с низкой активностью WNT создается с помощью сигналов, исходящих из нервного гребня и градиента высокой активности BMP, исходящего из передней энтодермы (40).
Предыдущие исследования относительно передачи сигналов Wnt оказались противоречивыми, указывающими в основном на негативные эффекты секретируемых членов семейства Wnt или канонического пути Wnt на кардиогенез (40-47). Новые исследования подтвердили, что канонический путь Wnt позитивно регулирует кардиогенез стадио-специфическим образом и посредством взаимного общения между каноническим и не-каноническим путями Wnt, а также передачи сигналов Bmp: в ранних фазах он способствует детерминации в кардиогенный клон и супрессирует гаматопоэтический и сосудистый клоны, с противоположными эффектами на более поздних стадиях (48-50). Интересно, что этот эффект, по крайней мере, частично клеточно неавтономен, с зависимостью от секретируемых FGFs из соседней энтодермальной ткани и Sox 17, индуцируемого с помощью передачи сигналов Wnt (51-53).
Семейство генов Id состоит из 4-х членов, которые являются доминантно-негативными антагонистами basic helix-loop-helix транскрипционных факторов и регулируют дифференцировку во множественных клонах. Они экспрессируются исключительно в эпикарде, но не в миокарде и осуществляют свои функции исключительно посредством передачи сигналов от эпикарда к миокарду. Компаундные гетерозиготы по мутантным Id аллелям вызывают истончение миокардиальной стенки, межжелудочковой перегородки, эндокардиальных подушек и дефекты тракта оттока. В исследовании Fraidenraich et al. предоставляется несколько линий доказательств, что секретируемые факторы могут устранять мутантные Id фенотипы (54): как инъекционные Id мутантные бластоцисты с дикого типа embryonic stem (ES) клетками, так и инъекции ES клеток в перитонеум самок до оплодотворения устраняет эмбриональную летальность кардиального фенотипа, вызываемого потерей Id генов. Дальнейший анализ идентифицировал IGF-1 и Wnt5a в качестве наиболее вероятных, клеточно неавтономных, секретируемых факторов спасения. Wnt5a оказался способным нормализовать разрегулированные профили генной экспрессии в эксплантированных, культивируемых Id1-/-Id3-/- сердцах. Значение этих исследований двойное: эмбриональная летальность уродств может быть устранена с помощью секретируемых факторов, а эмбриональные ES клетки могут составлять ценный ресурс для идентификации секретируемых факторов восстановления.
Менее изученным сигнальным путем в кардиогенезе является путь planar cell polarity (PCP). Впервые идентифицированный в работах с
Drosophila, этот процесс означает установление оси полярности перпендикулярно апикально-базолатеральной оси и является важным для направленного движения клеточных популяций. Некоторые мутанты гомологов этого пути у млекопитающих вызывают нарушения тракта оттока и коронарных артерий (55, 56). Некоторые рецепторы этого пути коэкспрессируются в развивающейся сердечно-сосудистой системе и взаимодействуют генетически, чтобы давать мутантные фенотипы, очень напоминающие разные формы CHD людей (56, 57), это открывает возможность того, что компаундные гетерозиготы по мутациям пути PCP могут вызывать заболевания у людей (Fig. 5). Интересно, что Wnt5a недавно был идентифицирован в качестве важного кандидата на роль регулятора PCP путей у млекопитающих (58).
No flow, no grow: the role of hemodynamics as an epigenetic factor in heart development
Кардиальные нарушения у плодов обычно ассоциируют с аномалиями кровотока на уровне самого сердца, сосудов или сосудистой сети. Гипотеза 'no flow-no grow', часто используемая детскими кардиологами, гласит, что величина кровотока через структуру предопределяет степень её развития. Естественная история CHD во время плодной жизни, показывает, что некоторые формы болезни, в частности, аортальный стеноз, могут ухудшаться со временем к моменту развития hypoplastic left heart syndrome (HLHS) [for review, see (59)]. Как следствие, вмешательства на плодах помогают в смысле уменьшения любой гемодинамической компоненты пороков сердца. паттерн кровотока в развивающемся сердце, как уже давно было предположено, играет существенную роль в кардиальном морфогенезе. in the developing heart has long been proposed to play a significant role in cardiac morphogenesis. Спустя некоторое время стало известно, что альтерации кровотока внутри сердца у эмбрионов кур могут вызывать кардиальные и сосудистые пороки. Напр., перевязка правлой латеральной вителлиновой вены индуцирует субаортальные дефекты межжелудочковой перегородки и аномалии клапанов и сосудов (60), в то время как перевязка левого предсердия может давать фенотип, сходный с HLHS (61). Дальнейший анализ этого фенотипа показал
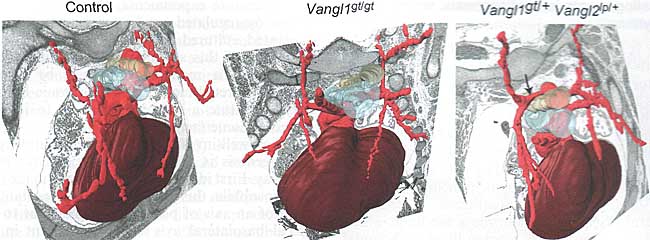 |
Fig. 5. Interaction of genetic components of the PCP pathway. Three-dimensional reconstruction of the arteries (bright red) of E14.5 wildtype mouse embryo (left), Vangllg'lg' (middle) and Vangllg'l+;Vangl2lp +hearts (right). The arrow denotes an aberrant right subclavian artery crossing behind the esophagus, which is not seen in Vang!lg'gl or Vangl2,p * animals, highlighting the genetic interaction between those two genes. Light blue = trachea, light yellow = esophagus.
|
Fig. 5. Interaction of genetic components of the PCP pathway. Three-dimensional reconstruction of the arteries (bright red) of E14.5 wildtype mouse embryo (left), Vangllg'lg' (middle) and Vangllg'l+;Vangl2lp +hearts (right). The arrow denotes an aberrant right subclavian artery crossing behind the esophagus, which is not seen in Vang!lg'gl or Vangl2,p * animals, highlighting the genetic interaction between those two genes. Light blue = trachea, light yellow = esophagus.
четкое сходство аномалий трехстворчатого и митрального клапанов с обнаруживаемыми у людей HLHS (62), а также задержку созревания системы His-Purkinje (63), возможно в результате изменений в регуляции молекул, зависимых от shear стрессов, таких как Etl и Nrgl. У рыбок данио количественное отображение
in vivo обнаруживает высокий сдирающий (shear) стресс, вызываемый вихревым током в развивающемся сердце. После закрытия кровотока или на уровне трактов кардиального притока или оттока, сердца с аномальными третьими камерами, уменьшают петлеобразование и нарушают образование клапанов (64). Эти наблюдения указывают на то, что сдвигающие (shear) силы, генерируемые кровотоком, не только регулируют гемодинамику, но и онтогенетические реакции. Экспрессия KLF2 отражает эти внутрикардиальные сдвигающие силы, а специфичная для эндотелия делеция этого гена ведет к летальной сердечной недостаточности из-за состояния высокого минутного сердечного выброса (65).
Недавнее исследование асимметрии развития аортальной дуги проливает свет на иерархию генетических в противоположность гемодинамическим причинам неправильного развития аортальной дуги (66). У мутантных мышей, у которых отсутствует обычная асимметричная экспрессия
Pitx2 на левой стороне тракта оттока, изучалось аномальное ремоделирование branchial arch arteries (BAAs). После исключения того, что это может быть обусловлено прямыми эффектами
Pitx2-позитивных клеток вблизи BAAs, авт. установили, что аномальная ротация тракта оттока у мутантных мышей индуцирует неравномерное распределение кровотока в шестую BAA, нарушая тем самым передачу сигналов посредством PDGFR/VEGFR. Эти результаты в дальнейшем были подтверждены с использованием микрохирургических и фармакологических подходов: перевязка левой шестой BAA ведет к персистенции правой шестой BAA, а блокирование PDGFR и VEGFR индуцирует двухстороннюю потерю шестых BAA. Эти результаты привели авт. к предположению событий домино-подобного каскада, при котором генетические и гемодинамические части перемежаются при формировании нормального или аномального развития.
Congenital heart defects in humans: arguments for a genetic basis
Generally. CHD, как полагают, возникает спорадически, а не на семейной основе в большинстве случаев. Кстати, исследование Baltimore-Washington остается наиболее обширным по охвату в полевых условиях (2). Общим знаменателем этого и др. исследований является то, что увеличение риска повторного появления намного выше при горизонтальной передаче (между детьми одних родителей) в противоположность вертикальной передаче (от родителей детям) (2. 67. 68). Причины этого феномена неизвестны. После подсчетов стратифицированных рисков повторного появления для специфических патологий, некоторые новые исследования стремятся теперь оценить количественно вклады генетических факторов в определенные болезни.
Анализ наследования выступает в качестве мощного инструмента для таких исследований и был использован в исследованиях по генетике левосторонних, соответствующих спектру болезней от BAV до аортального стеноза при HLHS. Наследуемость была подсчитана для BAV в отдельности в h2 = 0.99 и для BAV с др. CHD в h2 = 0.75 (69). Сходные результаты были получены в др. исследованиях, изучавших спектр обструкций тракта оттока из левого желудочка (70) или особенно HLHS (71).
Оба сегрегационных анализа, также как и исследования по сцеплению строго подтверждают, что некоторые локусы или модификаторы вносят вклад в болезнь в соответствии с др. исследованиями, рассматривающими CHD как сложный признак с образованием семейных кластеров, но часто без строгого соблюдения Менделевского наследования (72, 73). Интересно, что генетические исследования Keeshond модели конотрункальных дефектов сердца свидетельствуют в пользу трех взаимодействующих локусов (74), самый сильный из которых полностью перекрывается с одиночным локусом основателем, определенным у собак, моделирующих пороки трехстворчатого клапана (75). Вполне возможно, что эти результаты аналогичны патогенезу болезни Hirschsprung, с мутациями относительно ограниченного числа генов, вызывающих сложные паттерны доминантного, рецессивного и полигенного наследования.
Putting one plus one together: from candidate gene studies to whole genome association
Какрй инструмент может быть использован для обнаружения, какой из генетических дефектов вызывает фенотип у данного пациента? Главной опорой генетического анализа в последние два десятка лет по этой причине был анализ генов кандидатов и сцепления. Мы можем ожидать, что их клиническое использование д. увеличиваться, т.к. алгоритмы генетического тестирования начинают появляться (3) параллельно с увеличением мощи техник секвенирования и генотипирования.
Секвенирование генов кандидатов привело к лучшему определению генотип-фенотипических корреляций в особенности для Nkx2.5 (16-19) и GATA4 (79-82). В то время как носители мутации Nkx2.5 обнаруживают электрофизиологические нарушения в ассоциации с тетрадой Fallot и дефекты межпредсердной перегородки, GATA4 мутации вызывают дефекты перегородок и эндокардиальных подушек без атриовентрикулярного блока. При экстраполяции известных функций Tbx20 (see above), Kirk et al. показали, что мутации в этом гене вызывают разрушение T-box ДНК-связывающего мотива и вызывают семейные формы дефектов перегородки при их образовании, роста камер и васкулогенеза (83). Dilated cardiomyopathy , обнаруживаемая у взрослых пациентов, сходна с описанными у мышей фенотипами. В др. недавнем исследовании, проверявшем роль GATA4, который является транскрипционным кофактором, чья потеря у мышей вызывает дефекты межпредсердной перегородки, дефекты межжжелудочковой перегородки, конотрункальные и внесердечные аномалии. Когда были скринированы 392 пациентов с различными не-синдромальными CHD, то были найдены 3 missense мутации (84). Важная фенотипическая изменчивость была таже описана для человеческих GDF1 мутаций с дефектами от тетрады Фалло до d-транспозиции магистральных артерий, дефекты ранее рассматривающиеся как самостоятельная этиологическая категория, на базе чисто морфологических наблюдений (85). Кстати, такое исследование идентифицировало единственную фракцию CHD пациентов в качестве носителей мутации и это ставит задачу определения, как эти мутации связаны с функцией генов известных онтогенетических путей и с хорошо известными фенотипами у людей.
В последние годы исследования сцепления идентифицировали новый причинные гены, в частности, для aortic aneurysm (smooth muscle alpha-actin, Acta!) (86), aortic aneurysm with persistent ductus arteriosus (smooth muscle cell-specific myosin heavy chain, Myhll) (87), atrial septal defect without cardiomyopathy (alpha cardiac actin, Actcl) (88), and aortic stenosis (Notchl) (89). Современные исследования имеют целью определить спектр аллелей эти х генов в отобранной популяции пациентов и использовать знания о новых путях механистически выделенного молекулярного патогенеза дефектов перегородок и клапанов. Напр., Notchl физически взаимодействует с Runx2 , чтобы репрессировать его транскрипционную активность, а потеря его функции может приводить к Runx2-зависимой кальцификации створок клапанов (89). Также можно ожидать, что др. гены внутри контрактильных единиц гладкомышечных клеток, их места закрепления на внеклеточном матриксе, а также передача ими сигналов и транскрипционный аппарат должны приводить к болезням при родственных нарушениях. Наиболее поразительным является открытие пути кандидата безусловно объясняющего молекулярные основы синдрома Noonan и родственных нарушений [reviewed in detail in (90)]. Базируясь на инициальной идентификации SHP2 в качестве причинного гена посредством анализа сцепления (91), был идентифицирован RAS/MAPK какскад в качестве пути кандидата и был предположено участие, по крайней мере, 7 др. генов в синдроме Noonan и родственных нарушениях.
С появлением мощной технологии быстрого типирования полиморфизма одиночных нуклеотидов, исследования ассоциаций по всему геному, был найден способ их использования для изучения более распространенных болезней. Атриальная фибрилляция, общая аритмия, которая в основном возникает благодаря эктопической электрической активности около пульмональных вен, является важным фактором риска для паралича и сердечно-сосудистой гибели. В большом исследовании генетического варианта обеспечивающего риск возникновения фибрилляции предсердий во множественных этнически различающихся популяциях была идентифицирована интронная область Pitx2 (92). Эта работа, по-видимому, идентифицирует важный путь формирования лево-правосторонней оси тела и пульмональной вены (93) в качестве главного детерминанта персистенции или развития аритмогенных фокусов, отвечающих за атриальную фибрилляцию.
Эти исследования начинают выявлять аллельный спектр, лежащий в основе пороков сердца и их результаты внесут львиную долю в будущий учебник CHD, подразделенный на главы, посвященные генетическим путям.
Chromosomal aberrations and copy number variations: a shortcut into the developmental underpinnings of CHD
Задолго до открытия трисомии 21 в качестве генетической причины синдрома Дауна, была распознана ассоциация дефектов типов атриовентрикулярного канала (94). С тех пор были описаны многочисленные ассоциации между хромосомными аберрациями и CHD [for review, see (3)], а техника криотипирования была развита от микроскопической техники высокого разрешения до высоко-производительной детекции copy number variations (CNVs) на уровне в 10 kb.
В недавнем исследовании Thienpont et al. использовали набор бактериальных искусственных хромосом в кагорте из 60 пациентов с CHD , с несиндромальным паттерном дисморфических признаков или умственной отсталостью, из которой они исключили известные генетические нарушения на базе дисморфологии и анализа высокого разрешения G-дисков (95). Базируясь на строгих критериях, хромосомные аберрации были найдены в качестве этиологических для CHD в 17% случаев, это сравнимо с долей их у пациентов в умственной отсталостью. Даже если принять во внимание, что CNVs явдя.тся более частым источником вредных геномных вариаций, затрагивающих 12% генома (96), то это исследование указывает на то, что новая базирующаяся на массивах техника несет выдающиеся перспективы для диагностического оснащениe и является наиболее рациональным способом для целенаправленного поиска новых генов, вызывающих CHD. В качестве аналогии, детекция аномалий кариотипа послужила инструментом для открытия PROSIT240, участвующего в несиндромных случаях d-транспозиции крупных артерий, а CHD7, основной причиной синдрома CHARGE (97, 98).
Somatic mutations: similarities between malformations, adult-onset heart disease and cancer?
Считается, что соматические мутации являются важными для патогенеза рака. Идея одиночной соматической клетки, подвергающейся мутации и затем вносящей вклад в порок сердца является привлекательной в свете клональной природы экспансии клеток в сердце (15). В то же самое время соматические мутации д.быть совместимы с наблюдением. что большинство случаев CHD происходит на спорадической основе и что сердце является единственным затрагиваемым органом (99). В недавних исследованиях archival тканей сердца были найдены мутации нескольких ключевых регуляторов развития сердца в избранных регионах исследованных сердец, но не в контроле (100-102). Мутационный скрининг генов не связанных с развитием сердца не выявил каких-либо достоверных изменений последовательностей. Сходное исследование с использованием нефиксированных тканей от пациентов с болезнями аорты не выявили каких-либо изменений последовательностей в гене
Nkx2.5 (103). Gallob et al., проводили скрининг на GJA5, который кодирует белок щелевых соединений Connexin 40, и является кандидатом на роль гена фибрилляций предсердий, в ДНК, экстрагированной из предсердий и нашли 4 мутации у 15 пациентов. В 3-х из 4-х случаев этих мутации не были обнаружены в ДНК лимфоцитов, подчеркивая соматический мозаицизм, тогда как в одном случае, присутствие мутации указывает на передачу с зародышевой линией (104). Эти исследования ожидают дальнейшего подтверждения независимыми исследователями.
Leave no gene behind: perspective
В последние 50 лет диагностика и терапия в педиатрической кардиологии достигли впечатляющего успеха за счет исключительно описательного анатомического подхода, но редко в связи с информацией о молекулярном патогенезе. В последние годы многочисленные новые кардиальные регуляторы и морфогенетические механизмы были идентифицированы на молекулярном уровне, это позволило связать эти два подхода. Детальное молекулярное описание различных кардиальных полей и ballooning модели формирования камер предоставило новую основу для понимания морфологии CHD. Известно : генетические детерминанты в этом процессе связывают очень убедительно континуум болезни от пороков до аритмий и кардиальной дистрофии/дисфункции, это дает клиницистам объяснение фенотипов, которые были трудны для понимания, исходя только из гемодинамических или морфологических подходов. Мы начинаем понимать, напр., почему в одной семье ребенок имеет дефект перегородки, а один из родителей пользуется электрокардиостимулятором. Наиболее вероятным сценарием является то, что множественные генетические и эпигенетические факторы взаимодействуют у данного пациента с CHD, объясняя сложный характер признаков, наблюдаемый в большинстве случаев. Согласно аналогичным исследованиям, проведенным в др. области, напр., во взрослой кардиологии или аутизме, ключевые рессурсы д. разрабатываться для педиатрических сердечно-сосудистых болезней более крупными много-отраслевыми кагортами. Такие кагорты смогут предоставить нам уникальную возможность транслировать информацию основных наук на риск stratification и уход за пациентами, исходя из причины болезни.
Сайт создан в системе
uCoz