The ups and downs of holoprosencephaly: dorsal versus ventral patterning forces
 Clin Genet Volume 73 Issue 5, Pages 413 - 423, 2008, В© Blackwell Munksgaard, 2008. | |
|
Голопрозэнцефалия (НРЕ), характеризующаяся неполным разделением переднего мозга и лицевых компонентов на левую и правую половины, является результатом распространенного дефекта развития у человека. Причины заболевания - генетические и средовые факторы. Тяжесть фенотипических проявлений заболевания значительно варьирует. Генетические взаимодействия, лежащие в основе наследственных форм НРЕ сложны и пока не совсем ясны. Модельные животные, особенно мутантные мыши, значительно улучшили наше понимание того как идет развитие переднего мозга и как мозговые полусферы разделяются на правую и левую половины. Эти находки, а также характеристика некоторых генов, определяющих НРЕ, подтверждают, что в основе классической НРЕ и midline interhemispheric (MIH) лежат два механизма. Нарушение (прямое или опосредованное) вентрализирующего эффекта sonic hedgehog сигналинга является основным по отношению ко всем или к большинству форм классической НРЕ. А нарушение дорсализирующего эффекта передачи сигналов bone morphogenetic protein (BMP) может быть ключевым при MIH HPE.
|
Голопрозэнцефалия (Holoprosencephaly - HPE) - наиболее часто встречающийся у человека дефект развития переднего мозга. Эта аномалия развития встречается с частотой 1 на 5-10 тыс. новорожденных и 1 на 200-250 случаев при спонтанных абортах (1-5). Типичными признаками НРЕ являются: неполное разделение передней части переднего мозга или конечного мозга (мозгового пузыря) на левое и правое полушария, что в норме происходит между 18 и 28 днем внутриутробного созревания плода. Этот порок развития обусловлен дефектом формирования структур срединной линии. HPЕ как правило ассоциируется с черепно-лицевыми аномалиями. Однако в данном обзоре рассматриваются главным образом аномалии развития переднего мозга. Этиология НРЕ крайне гетерогенна, что является следствием влияния как средовых, так и генетических факторов, а также взаимодействия между ними (6-8). Широкий спектр НРЕ фенотипов может наблюдаться внутри одной семьи, где лица, несущие идентифицированную НРЕ мутацию, могут быть клинически нормальными или иметь тяжелые поражения (9-11). Такая крайняя гетерогенность НРЕ при семейных и спорадических случаях в настоящее время создает значительные трудности в установлении четких гено-фенотипических корреляций и при генетическом консультировании родителей, являющихся носителями НРЕ мутации, в отношении риска заболевания их потомства. Большинство сведений о генетических путях развития переднего мозга получено в основном при исследованиях на экспериментально-биологичеких моделях. Все увеличивающееся число мутантных моделей (мышей) с дефектами развития срединной линии мозгового пузыря мимикрирующими НРЕ человека дает возможность проникнуть в "онтологию" НРЕ человека и поднять новые вопросы, столь необходимые для более полного понимания этого крайне тяжелого врожденного дефекта. HPE: two classes and four types Основные сведения о HPE у человека почерпнуты после появления методов магнито-ядерного изображения и высококачественной X-ray компьютерной томографии. В результате анализа данными методами было выделено два основных класса НРЕ, охватывающих 4 типа заболевания. К этим двум классам относятся "классические" НРЕ, при которых наиболее тяжело пораженными областями полусфер являются базально/вентральный передний мозг и НРЕ межполушарнйо срединной линии (midline interhemispheric HPE - MIH HPE) или syntelencephaly (middle interhemispheric fusion variant (MIHF) or syntelencephaly) , при которой кортикально/дорсальная часть полусфер не разделена, но базальный передний мозг может оставаться нормальным (8, 12-14). "Классическая" НРЕ подразделяется на три различных типа. Наиболее тяжелыми фенотипическими проявлениями аномалий развития характеризуется алобарная НРЕ (алобарная голопрозэнцефалия развивается вследствие нарушения деления переднего мозга), при которой весь передний мозг является моновентрикулярным и уменьшенным в размере. Для этого типа характерна циклопия. При втором типе - полулобарной НРЕ - передние области полусфер не разделены, но задние области часто нормальны. Третий желудочек небольшого размера и частично сформирован, а кора, базальные ганглии и таламус аномально соединены. И, наконец, при лобарной НРЕ антериальная часть переднего мозга неполностью разделена, но в меньшей степени, чем при полулобароной НРЕ. Больные имеют полностью сформированный третий желудочек, хотя дисморфии и передние структуры (мозолистое тело и обонятельные луковицы) могут отсутствовать или быть гипопластичными. 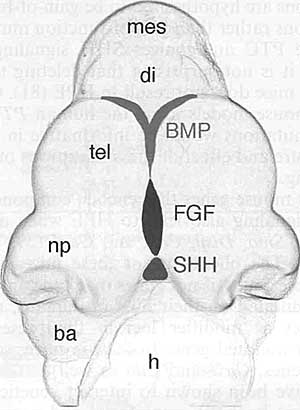 Fig. 1. Schematic representation of an E10.5 mouse head (frontal view, dorsal up). Telencephalic midline areas are highlighted in color, with the dorsal midline expressing bone morphogenetic proteins (red), the rostral midline expressing fibroblast growth factors (blue), and the ventral midline expressing sonic hedgehog (green), ba: branchial arch, di: diencephalon, h: heart, mes: mesencephalon, np: nasal process, tel: telencephalon . Fig. 1. Schematic representation of an E10.5 mouse head (frontal view, dorsal up). Telencephalic midline areas are highlighted in color, with the dorsal midline expressing bone morphogenetic proteins (red), the rostral midline expressing fibroblast growth factors (blue), and the ventral midline expressing sonic hedgehog (green), ba: branchial arch, di: diencephalon, h: heart, mes: mesencephalon, np: nasal process, tel: telencephalon . Второй класс НРЕ, известный как MIH HPE встречается реже и более мягкий форме по своим клиническим проявлениям в сравнении с классической НРЕ. В некоторых случаях наблюдают лишь поражения дорсального переднего мозга. Дорсальная часть полусфер не разделена на заднюю фронтальную и париетальную области и во многих случаях каудальные ядра и таламические структуры также не полностью разделены. Но в то же время имеется межполушарное разделение базального переднего мозга, передней части фронтальных долей и затылочных областей. Было установлено, что генетические пути, определяющие нормальное развитие дорсального переднего мозга, нарушены при MIH HPE, тогда как при классической НРЕ генетические пути, определяющие развитие вентрального переднего мозга, поражаются чаще (15). Genetic heterogeneity of HPE Генетика НРЕ сложна - было идентифицировано лишь несколько мутантных генов, определяющих семейные случаи НРЕ. До 45% больных НРЕ имеют выраженные цитогенетические аномалии - трисомию 13, трисомию 18 и триплоидию (16). Согласно кариотипическому анализу, описано, по меньшей мере, 12 геномных участков распределенных по 11 разным хромосомам (локусы НРЕ1- НРЕ 12), т.е. локусов, содержащих НРЕ кандидатные гены (8,17). Наследование может быть аутосомно-доминантным, рецессивным и сцепленным с Х-хромосомой (17, 18). Кроме того, НРЕ иногда ассоциируется с другими врожденными заболеваниями - синдромом Смита-Лемли-Опитца и синдромом Pallister-Hall (19, 20). Гетерогенность при семейных случаях НРЕ (от крайне тяжелых поражений до клинической нормы) с одинаковыми мутациями может быть обусловлена влиянием внешних или тератогенных факторов [например, алкоголем, диабетом, холестеролом, ретиноевой кислотой (21-23)] или генами-модификаторами (24). В соответствии с присутствием генов-модификаторов и взаимодействующих локусов, гетерогенные мутации в двух НРЕ генах должны приводить к тяжелому фенотипу. К настоящему времени описано три таких случая [sonic hedgehog (SHH) и TGIF (25), SHH и ZIC2 (25), и PTC1 и GLI2 (26)]. Это число, вероятно, будет увеличиваться по мере выявления кандидатных генов и понимания их взаимодействия. Для решения этой проблемы необходимы исследования на модельных объектах (мышах). Здесь следует заметить, что у мышей, в отличие от человека, гетерозиготная мутация одного гена, ассоциирующаяся с НРЕ, обычно не проявляется фенотипически. Аномальный фенотип наблюдают лишь у гомозиготных мутантов (Табл.1). Это свидетельствует о том, что мыши менее подвержены гаплонедостаточности по сравнению с человеком. Тем не менее, гены, ассоциированные с НРЕ у человека и гомозиготные у мышей, чаще приводят к НРЕ. Мышиные мутанты с НРЕ фенотипом (гены которых пока не связывают с НРЕ человека), могут способствовать обнаружению новых генов-кандидатов и генов-модификаторов при НРЕ. Другим преимуществом моделей является то, что мутации в двух или более генах могут быть довольно быстро скомбинированы для изучения генетических взаимодействий, приводящих к НРЕ (Табл.1). Несколько Таблица 1  генов, которые спонтанно мутировали у мышей [например, Cdo и Tgif (27-29)] вели к НРЕ с вариабельной экспрессивностью или пенетрантностью, мимикрируя признаки, наблюдаемые при НРЕ человека. Например, у Cdo мутантов разброс фенотипических отклонений был довольно велик - от нормального переднего мозга до полулобарной НРЕ. Спектр вариабельности фенотипических отклонений у этих мутантных мышей зависел от генетической линии животных, что говорит о контроле гетерогенности генами-модификаторами (28-30). Вероятно, идентифицировать эти гены можно используя анализ рекомбинантных инбредных линии (quantitative trait loci analyses). Генетические взаимодействия, лежащие в основе гетерогенности НРЕ фенотипа у человека, только сейчас начали поддаваться пониманию. И мышиные модели будут в ближайшие годы главным объектом для выявления генетических путей, регулирующих формирование средней линии конечного мозга. Development of the telencephalic midline Передний мозг у млекопитающих развивается из эмбрионального прозэнцефалона, локализованного в наиболее передней части нервной трубки. Вскоре после закрытия нервной трубки (примерно на 9,5 день у мышей и на 3,5 неделе внутриутробного развития у человека) прозэнцефалон начинает дифференцироваться на конечный мозг (telencephalon) - будущие полусферы мозга, и диэнцефалон - будущий таламус. К Е10.5 у мышей и 35 день у человека конечный мозг подвергается значительным морфологическим изменениям, начиная расщепляться медиально на два билатеральных пузыря (Рис.1). В отличие от клеток расположенных более латерально, клетки срединной линии имеют более высокую гибель и сниженную пролиферацию, приводя к разделению расширяющегося мозга на правое и левое полушария. Клетки дорсальной срединной линии дифференцируются медиально в хороидное сплетение, секретирующее цереброспинальную жидкость, и в прилегающую кортикальную кайму, которая индуцирует формирование гиппокампа и является также источником клеток Кахаля-Ретциуса (31-34). Вентрально и рострально клетки срединной линии способствуют образованию септума и ганглиозного бугорка, которые дают начало компонентам базального ганглия. Несколько секретируемых сигнальных молекул экспрессируются телэнцефалическими клетками срединной линии. Дорсально срединная линия экспрессирует bone morphogenetic proteins (BMPs) и wingless-Int proteins (WNTs), а ростральная и вентральная срединная линия экспрессируют fibroblast growth factors (FGFs) и SHH соответственно (Рис.1). Предполагается, что эти факторы взаимодействуют при формировании и паттернировании полусфер конечного мозга (35-37). The dorsal midline Для развития дорсальной средней линии необходимы BMP сигналы. По меньшей мере 5 DVH генов экспрессируются в дорсальной средней линии [Bmp2. Bmp4, Bmp5, Bmp6, and Bmp7; (38)]. BMP4-пропитанная гранула, помещенная в культуру эксплантанта латерального конечного мозга, индуцировала признаки дорсальной средней линии - клеточную гибель, низкий уровень пролиферации, экспрессию маркера средней линии Msxl и репрессию маркера Foxgl не характерного для средней линии (38). 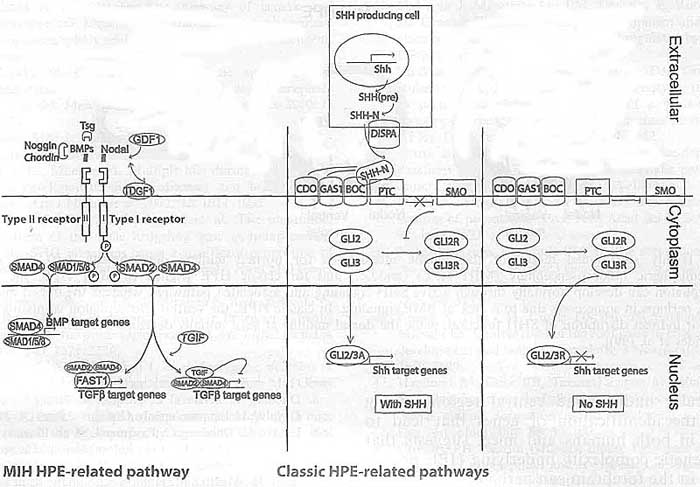 Fig.2. Schematic representation of the pathways involved in MIH (blue) and classic (orange) holoprosencephaly in humans and mice [Adapted from Ming and Muenke (24) and Krauss (30)]. BMP, bone morphogenetic protein. Специфичный для конечного мозга нокаут ВМР рецепторного гена типа I показал, что сигнал ВМР требуется для формирования дорсальной средней линии (39, 40). Следует заметить, что у двойных Bmprla и Bmprlb мутантов конечный мозг не разделен на левую и правую полусферы, что мимикрирует MIH BMP (midline interhemispheric HPE) (40). Однако у человека пока не определена роль разрушения сигнала ВМР при НРЕ. Кроме ВМР генов, гены WNT - Wnt2b, Wnt3a, Wnt5a и Wnt8b - экспрессируются в дорсальной средней линии (31, 32). И хотя Wnt3a важен для развития гиппокампа (32, 41), WNTs не участвует в формировании средней линии. Однако нокаут Rfx4-v3 (транскрипционный вариант, кодирующий winged-helix transcription фактор), ведет к утрате Wnt3a экспрессии и НРЕ (42). Тем не менее, не установлено прямого участия WNT генов в формировании дорсальной средней линии и в НРЕ. В настоящее время известно, что кроме ВМР рецепторных генов, мутации только двух других генов вызывают утрату дорсальной средней линии - Fgf8 и Zic2. И в обоих случаях эти мутации являются гипоморфными (43,44). Zic2 кодирует транскрипционный фактор цинковые пальцы, экспрессируемый в дорсальной и вентральной средней линии (40,45). Экспрессия Zic2 не зависит от BMP сигналов (40), но может быть индуцирована эктопической FGF8 аппликацией (47), подтверждая тем самым, что он действует ниже (downstream) FGFs и параллельно или выше (upstream) BMPs (Рис.3). Хотя вентральный конечный мозг, который оказался нормальным у Zic2 мутанта, не был тщательно изучен (44), ZIC2 мутации у человека могут вести как к классической, так и к MIH HPE (см. ниже) (15). 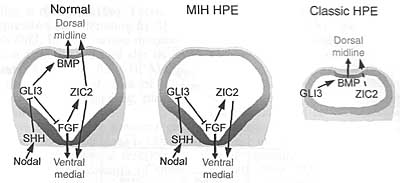 Fig. 3. Highly streamlined model of the genetic interactions for normal midline development (left), for midline interhemispheric holoprosencephaly (MIH HPE) (middle), and for classic HPE (right). In MIH HPE. the ventral telencephalon can develop normally through active SHH signaling and associated pathways, whereas the dorsal midline is absent, perhaps in some cases due to a lack of BMP signaling. In classic HPE, the ventral telencephalon is missing due to direct or indirect disruption of SHH function, while the dorsal midline at least initially develops normally [Adapted from Fernandes et al. (40)]. Fig. 3. Highly streamlined model of the genetic interactions for normal midline development (left), for midline interhemispheric holoprosencephaly (MIH HPE) (middle), and for classic HPE (right). In MIH HPE. the ventral telencephalon can develop normally through active SHH signaling and associated pathways, whereas the dorsal midline is absent, perhaps in some cases due to a lack of BMP signaling. In classic HPE, the ventral telencephalon is missing due to direct or indirect disruption of SHH function, while the dorsal midline at least initially develops normally [Adapted from Fernandes et al. (40)].The rostral midline Ростральная средняя линия экспрессирует 5 Fgf генов: Fgf3, Fgf8, Fgfl5 (Fgfl9 у человека), Fgf 17, и Fgf 18 (48-51). FGF8-пропитанные гранулы, помещенные в дорсо-латеральную область конечного мозга цыпленка, могут индуцировать эктопические извилины (бороздки) с признаками ростральной средней линии (52). Это означает, что FGFs могут играть определенную роль в формировании ростральной срединной линии. Однако никакие мутации в FGF пути пока еще не связывают с утратой ростральной средней линии у мышей и человека. The ventral telencephalon, including the midline Shh важен для развития вентрального переднего мозга - мутации этого гена у мышей и человека приводят к классическому фенотипу НРЕ (53-57). В переднем мозге Shh мышиных мутантов остаются только дорсальные предшественники, а все вентральные предшественники, которые в норме дают начало базальному ганглию, утрачены (53, 58-60). Наряду с ролью индуктора вентральных нервных клеток, Shh также необходим для поддержания пролиферации и выживаемости вентральных клеток-предшественников (54, 61-63). У Shh мутантов утрата вентральных структур сопровождается явным отсутствием разделения дорсальных полусфер конечного мозга (53, 63). Это, по всей вероятности, не обусловлено отсутствием дорсальной средней линии, а, скорее, обусловлено отсутствием общего роста и увеличения объема полусфер (40, 60). Утрата вентральных клеток, наблюдаемая у Shh мутантов, может быть предотвращена, если его нижестоящий антагонист GU3 также мутантен. Это говорит о том, что и другие факторы, а не только SHH, могут индуцировать вентральное развитие (58, 64). Известно, что FGF также необходим для данного процесса. У Fgfrl и Fgfr2 двойных мутантов или Fgf8 мутанта вентральные клетки не могут быть генерированы (43, 65). Более того, SHH генетически действует выше (upstream) FGFs при формировании вентрального конечного мозга. SHH не только регулирует экспрессию нескольких Fgf генов - Fgf3, Fgf8, Fgf 15, Fgf 17, и Fgfl8 (60, 63, 64), но также зависит от FGF сигналов, без которых не формируются все вентральные области, и. даже в тех случаях, когда SHH экспрессируется и проявляет активность, развития вентральных структур не наблюдают, если нарушен FGF сигналинг (65). И, напротив, сигналы FGF могут эктопически индуцировать вентральную генную экспрессию, даже в тех случая, когда нарушено SHH сигнализирование (66). Регуляция экспрессии FGF и передача сигналов посредством SHH косвенно осуществляются через GU3. У GU3 мышиных мутантов Fgf генная экспрессия увеличивается, а конечный мозг вентрализирован (31, 64, 66, 67). Более того, в отличие от Shh мутантов, утрата GU3 не предотвращает утрату FGF передачи сигналов, что свидетельствует о том, что FGFs находится ниже (downstream) GU3 (65). В настоящее время механизмы генетической регуляции развития средней линии мозга пока поняты недостаточно, но предыдущие исследования очерчивают рамки, в пределах которых есть смысл вести дальнейшие исследования, чтобы понять генетические пути формирования срединной линии мозга. SHH: the central player in classic HPE? Некоторые мышиные модели, мимикрирующие классические формы НРЕ, в настоящее время хорошо изучены. Все эти модели, также как и мутации человека, обусловливающие классические формы НРЕ, идентифицированы. И предполагается, что общим "знаменателем" в этих случаях является нарушение передачи SHH сигналов - прямо или косвенно, что ведет к утрате клеток вентрального типа. The SHH pathway SHH путь активно обсуждался как у позвоночных, так и у беспозвоночных (Рис.2 и обзор Fuccillo et al. and Chen et al. - 68, 69). В отсутствии внеклеточного SHH трансмембранный белок Smoothened (SMO) ингибируется SHH рецептором Patched (PTC). Связывание SHH с РТС облегчает ингибирование SMO и способствует функционированию GLI2 транскрипционного активатора, в то же время ингибируя репрессорную форму GLI13. Некоторые трансмембранные белки также способствуют сигналингу SHH. В SHH -продуцирующих клетках DispatchedA (DISPA) увеличивает количество разъединенных, активных форм SHH (SHH-N) (70), тогда как в SHH-responding клетках GAS1, CDO, и BOC по предположениям способствуют SHH сигналингу через РТС рецептор (29, 71-74). Mutations in the SHH pathway lead to classic HPE in mouse and human У человека классическую форму НРЕ определяют мутации трех генов, кодирующих компоненты SHH сигнального пути SHH (HPE3), PTC (HPE7), и GL12 (HPE9) (56, 57, 75-80). Делеция только одного из этих генов - Shh - ведет к НРЕ у мышей. Утрата мышиного GH2 не нарушает развития переднего мозга. РТС мутации у человека, по некоторым предположениям являются скорее мутациями gain-of-function, чем loss-of-function, т.к. РТС противодействует SHH сигналингу (75). Поэтому не удивительно, что делетирование Ptc гена у мышей не ведет к НРЕ (81). Создание мышиных моделей с использованием РТС миссенс-мутаций человека было бы крайне полезным для установления природы и эффектов этих мутаций на SHH сигналинг. Другие гены мышей, кодирующие компоненты SHH сигналинга, также ведут к НРЕ при их делециях. Это гены Smo, Disp, Cdo, и Gasl (29. 70. 71, 73, 82). Фенотип мутантных животных подтверждает, что эти гены могут действовать как модификаторные локусы (modifier loci) в присутствии других мутантных генов. Фактически, у мышей некоторые из этих генов - Gasl и Cdo так же как и Gasl и Sh, взаимодействуют генетически и усугубляют, по крайней мере, черепно-лицевые дефекты и дефекты нервной трубки (72, 73). Cdo мутанты демонстрируют широкий спектр фенотипов в зависимости от генетической линии животных, что указывает на присутствие еще неидентифицированных модификаторных локусов (28, 29). В настоящее время у человека известно три примера генетического взаимодействия между двумя локусами, приводящими к НРЕ: SHH и TGIF (25), SHH и ZIC2 (25), и GLI2 и PTC (26). Disruption of other pathways that modulate SHH signaling results in HPE Гены мышей, косвенным образом модулирующие SHH сигналинг, при возникновении в них мутаций, могут приводить к НРЕ фенотипу. К примеру, мыши, дефицитные по генам, ингибирующим BMP активность, - Noggin и Chordin и MegalinjLrp2, имеют пониженный SHH сигналинг и утрату клеток вентрального типа, что напоминает классическую НРЕ (83,84). Другим примером являются компоненты FGF сигнального пути. Как было показано, у Fgf8 и Fgfrl;Fgfr2 мутантов, FGF сигналинг, подобно SHH, нужен для генерирования клеток вентрального типа в конечном мозге (43, 65), а SHH зависит в данном процессе от FGFs (65). Исследования на этих мышах позволило выделить дополнительные кандидатные гены, ассоциированные с классической формой НРЕ человека. Мутирующие компоненты Nodal сигнального пути (РИС.2) также ассоциируются с НРЕ человека и мышей. Утрата Nodal сигналов вела к утрате Shh экспрессии (85,86), свидетельствуя о том, что нарушения Nodal пути косвенным образом приводит к НРЕ через утрату SHH сигналинга. У человека мутация в гене TDGF1 (87) (кодирующем внеклеточный кофактор, облегчающий Nodal связывание с его рецептором - FOXH1 (FASTI) (8, 12, 24)), которая кодирует транскрипционный фактор, способствующий экспрессии Nodal-responsive генов, и TGIF [HPE4, (88)], кодирующий транскрипционный кофактор, ингибирующий Nodal путь, уже связывают с НРЕ. Сходный механизм выявлен и у мышей - мутации в генах, связанных с Nodal сигналингом - сам Nodal и Nodal с его внеклеточным кофакторным геном Gdfl, его рецепторным геном ActrHA или его нижестоящим эффекторным геном Smad2, могут приводить к фенотипам с признаками классической НРЕ (86, 89-91) (Table 1). Таблица 2 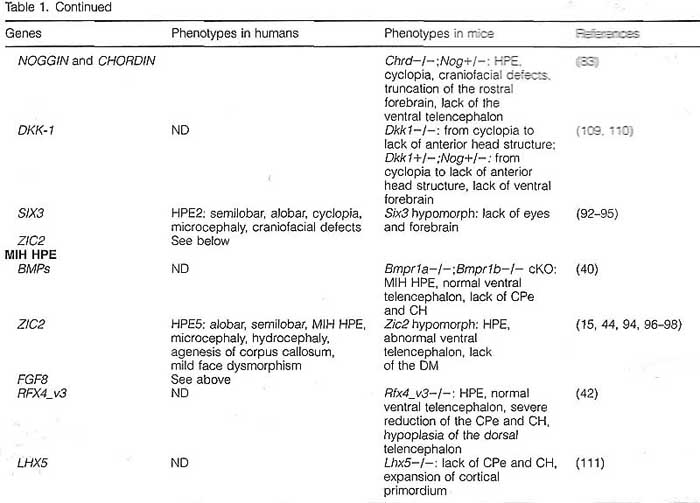 Six3 кодирует гомеодоменный транскрипционный фактор семейства Six/sine oculis. У человека SIX3 картируется в HPE2 локусе (92-94). У мышей частичная утрата функций Six3 ведет к тотальному отсутствию глаз и переднего мозга (95), что затрудняет установление связи между между Six3, SHH сигналингом и развитием вентрального конечнгого мозга. Однако, вероятно, что даже небольшие мутации Six3 также ведут у потере SHH сигналинга и классическому НРЕ-подобному фенотипу у мышей. MIH HPE, a different molecular basis У человека Z1C2 (HPE5) чаще ассоциируеся с признаками классической HPE. Однако это пока единственный ген, мутации которого также ассоциируются с MIH HPE (15, 94, 96-98). У мышей гипоморфный аллель Zic2 определяет фенотип, имеющий значительное сходство с таковым у человека. Кроме Zic2 , только гены, связанные с BMP передачей сигналов, продемонстрировали MIH-подобный фенотип у мышей (Рис. 2 и 3). Особенно ярко выражены фенотипические аномалии у двойных мутантов Bmprla и Bmprlb - у них наблюдали утрату клеток, типичных для дорсальной средней линии, тогда как Shh экспрессия и вентральное развитие были нормальными (40). Фактически, молекулярные причины классической и MIH HPE могут быть оппозитными, поскольку BMP сигналинг и SHH сигналинг по всей невральной оси оказывают оппозитные, если не антагонистичные эффекты на дорсо-вентральное паттернирование. В соответствии с этим, мутации, ведущие к усилению BMP сигналинга - в MegalinjLrp2, Noggin и Chordin, нарушают SHH экспрессию и ведут к классическому HPE фенотипу (83, 84). И, напротив, увеличение количества активного SHH ведет к снижению генной экспрессии Bmp и появлению признаков MIH HPE (99). Perspective Понимание генетических путей, регулирующих развитие переднего мозга, и в особенности срединной линии и вентральных областей, наряду с идентификацией генов, ведущих к НРЕ у человека и модельных мышей. Подтверждает, что генетические причины, определяющие НРЕ фенотипы в переднем мозге могут, вероятно, быть достаточно простыми и ограничены сигнальными путями - теми, которые модулируют SHH вентрально и модулирующими BMP дорсально (Рис.3). Более того, исследования с использованием модельных животных, указывают на существование кандидатных генов у человека, способных действовать как модификаторы и быть ответственными за широкий спектр НРЕ фенотипов. Идетификация таких генов крайне важна, особенно для пренатальной диагностики и прогнозирования голопрозэнцефалии. |