Посещений: 
ВРОЖДЕННЫЕ БОЛЕЗНИ СЕРДЦА
Использование iPSCs для выяснения основ
Decoding Genetics of Congenital Heart Disease Using Patient-Derived Induced Pluripotent Stem Cells (iPSCs) Hui Lin1, Kim L. McBride, Vidu Garg and Ming-Tao ZhaoFront. Cell Dev. Biol., 21 January 2021 | https://doi.org/10.3389/fcell.2021.630069
| |
|
Congenital heart disease (CHD) ведущая причина врожденных дефектов, затрагивающая ~1% живорожденных в США (Hoffman and Kaplan, 2002; Nees and Chung, 2019). CHD характеризуется морфологическими аномалиями сердечных камер, перегородок и клапанов, а также магистральных сосудов, исходящих из сердца. Врожденные нарушения всех аспектов описаны и большинство распространенных типов CHD может быть отнесено к след. категориям: (1) дефекты сердечных перегородок, (2) конотрункальные и артерий аортальных дуг аномалии, (3) обструктивные дефекты правостороннего- и левостороннего тракта оттока и (4) лево-правосторонне аномалии (heterotaxy) (Garg, 2006; Bruneau, 2008). Дефекты перегородок состоят из atrial septal defects (ASD), ventricular septal defects (VSD) и atrioventricular septal defects (AVSD) , тогда как распространенные конотрункальные и аномалии артерий аортальных дуг включают тетраду Фалло (TOF), сохранение и прерывание дуг аорты. Обструктивные повреждения правостороннего тракта оттока включают стеноз легочного ствола и атрезию клапанов легочной ствола с интактной межжелудочковой перегордкой (PA-IVS), тогда как hypoplastic left heart syndrome (HLHS), aortic valve stenosis (AVS) и bicuspid aortic valve (BAV) являются распространенными обструктивными дефектами лево-стороннего тракта оттока. Аномалии лево-право-сторонней передачи сигналов у развивающихся эмбрионов затрагивают образование сердечной петли, это является критическим для собственно расположения предсердных камер в соответствии с их желудочками и магистральными сосудами. Такое нарушения соотв. передачи сигналов ассоциирует со сложными формами CHD, такими как двойной выход из правого желудочка и двойной вход в левый желудочек, клинически обозначаемые как синдром heterotaxy (Kathiriya and Srivastava, 2000). Др. крупные CHD, которые не согласуются с упомянутыми выше категориями включают изолированные аномалии клапанов (напр., Ebstein's anomaly трехстворчатого клапана и пролапс митрального клапана), тотальные аномалии соединения легочных вен, аномалии коронарных артерий и patent ductus arteriosus.
Эпидемиологические исследования выявили, что генетические факторы являются главной причиной CHD, тогда как внешне-средовые факторы (внешние воздействия, материнское состояние, внутриматочное окружение и т.д.) также вносят весомый вклад (Liu et al., 2013; Pierpont et al., 2018). В целом, специфические генетические и внешне-средовые факторы могут быть идентифицированы в 20-30% случаев CHD. Генетические механизмы, лежащие в основе возникновения CHD сложные и остаются пока неуловимыми (Liu et al., 2017; Pierpont et al., 2018). Имеется ограниченное количество животных моделей для изучения генетики CHD, а трансгенные мыши, несущие человеческие варианты, не всегда воспроизводят клинические фенотипы CHD (Majumdar et al., 2019). Человеческие iPSCs, получаемые из соматических клеток (таких как кожные фибробласты или моноядерные клетки периферической крови) обладают потенциалом генерировать все типы клеток тела, происходящие из трех зародышевых слоёв (Takahashi et al., 2007; Yu et al., 2007). В сравнении с животными моделями iPSCs пациентов являются клинически подходящими и также включают генетический фон затронутых индивидов зависимым от болезни способом, это предоставляет мощный инструмент для изучения вклада данного генетического варианта в CHD. Специфические для пациента iPSCs могут быть дифференцированы в кардиомиоциты, эндотелиальные и эндокардиальные клетки, в кардиальные фибробласты и гладкомышечные клетки, это облегчает исследование сложной генетической регуляции и взаимодействий между генами и средой одновременно во многих типах клеток сердца (Hu et al., 2016; Zhao et al., 2017a; Gifford et al., 2019). Недавние исследования продемонстрировали, что с отредактированным геномом iPSCs являются идеальными платформами для выяснения регуляторных ролей не кодирующих генетических вариантов для риска возникновения болезни коронарных артерий и для изучения вклада комбинаторных взаимодействий многих генетических вариантов в сложную сердечно-сосудистую болезнь (Lo Sardo et al., 2018; Deacon et al., 2019).
Genetics of CHD Благодаря успехам массивного параллельного секвенирования, генетика CHD активно исследовалась в последнюю декаду. Крупные научные усилия, такие как NIH-funded Pediatric Cardiac Genomics Consortium (PCGC) привели к становлению скоординированных исследований генетических вариантов, встречающихся при CHD (Pediatric Cardiac Genomics Consortium et al., 2013; Jin et al., 2017). Генетические основы CHD могут быть сгруппированы в две категории: синдромальные CHD и не синдромальны (изолированные) CHD (Pierpont et al., 2018). Синдромальные CHD определяются как CHD совместно с др. врожденными аномалиями, дефектами нейрального развития и/или признаками дизморфии. Синдромальные CHD могут быть вызваны анеуплоидией, вариантами количества копий (инверсии или делеции более 1000 нуклеотидов), или дефектами одиночных генов. Down syndrome (trisomy 21) является широко распространенной хромосомной аномалией и 40-50% таких пациентов имеет разного типа CHD, с дефектами кардиальной перегородки чаще всего. Синдром Turner вызывается полной или частичной потерей X хромосомы, а левосторонние дефекты (coarctation of the aorta, COA), BAV и HLHS присутствуют у 30% таких пациентов. Синдром делеции 22q11.2 является одним из наиболее распространенным среди вариантов числа копий с делециями более 40 генов на хромосоме 22. Дефекты тракта оттока присутствуют у 75-80% пациентов с делециями 22q11.2. Синдромальные CHD вызываются дефектами одиночных генов, включая синдром Alagille (варианты в JAG1 и NOTCH2) и синдром Holt-Oram (варианты в TBX5) (Basson et al., 1997; Li et al., 1997b; Table 1). Генетические вклады в изолированных CHD были выявлены в последние две декады и большинство вариантов оказалось расположенными в генах, которые участвуют в молекулярной регуляции кардиального развития. Синдромальные и изолированные CHD обладают отличающейся генетической архитектурой: de novo protein-truncating variants (PTVs) довольно часты при синдромальных CHD , тогда как врожденные PTVs в основном обнаруживаются у пациентов с изолированной CHD (Sifrim et al., 2016; Jin et al., 2017).
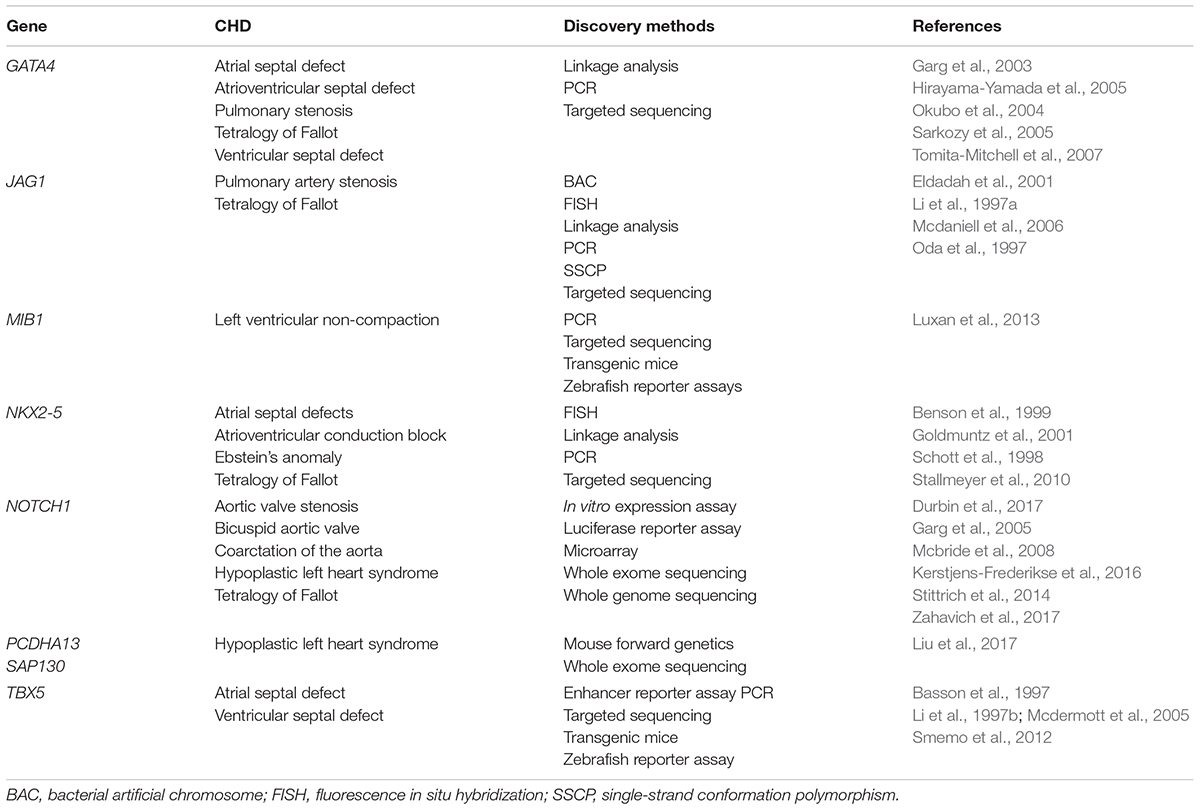 Table 1. A summary of single-gene variants underlying CHD.
Table 1. A summary of single-gene variants underlying CHD.
Патогенетические варианты, сцепленные с изолированными CHD, преимущественно кодируют транскрипционные факторы, сигнальные молекулы, структурные белки и эпигенетические модификаторы, которые важны для нормального развития сердца (Zaidi et al., 2013; Pierpont et al., 2018; Nees and Chung, 2019; Table 1). Напр., генетические варианты в высоко консервативных транскрипционных факторах являются критическим для кардиального развития и обнаруживаются как в семейных, так и спорадических случаях CHD. NKX2-5 варианты присутствуют у пациентов с TOF и ASD при задержке проводимости (Schott et al., 1998; Benson et al., 1999; Goldmuntz et al., 2001; Stallmeyer et al., 2010). Патогенные GATA4 варианты, ассоциированны с ASD, VSD, AVSD, pulmonary stenosis (PS) и TOF (Garg et al., 2003; Okubo et al., 2004; Hirayama-Yamada et al., 2005; Sarkozy et al., 2005; Tomita-Mitchell et al., 2007). Небольшой субнабор индуцированных GATA4 вариантами пороков сердца у людей воспроизведен у трансгенных модельных мышей, обладающих мутантной формой варианта GATA4 человека (Misra et al., 2012; Han et al., 2015).
Компоненты пути передачи сигналов NOTCH сцеплены с синдромальными и изолированными CHD. JAG1 варианты обнаруживаются у ~90% пациентов с синдромом Alagille, тогда как варианты NOTCH2 отвечают за дополнительные 1-2% индивидов с синдромом Alagille (Li et al., 1997a; Oda et al., 1997; Mcdaniell et al., 2006; Kamath et al., 2012). Варианты потери функции в JAG1 вызывают стеноз легочной артерии и TOF с или без атрезии пульмонального ствола (Eldadah et al., 2001). Гетерозиготные мутации DLL4 (лиганд) и NOTCH1 (рецептор) приводят к синдрому Adams Oliver с CHD, присутствующей примерно у 25% таких пациентов (Stittrich et al., 2014; Meester et al., 2015). Варианты RBPJ, которые взаимодействуют с расщепленным NOTCH1 белком, чтобы сформировать транскрипционный комплекс, также сцеплены с синдромом Adams Oliver (Hassed et al., 2012). Конечно, патогенные NOTCH1 мутации сцеплены с BAV, HLHS, AVS, COA и TOF (Garg et al., 2005; Mcbride et al., 2008; Kerstjens-Frederikse et al., 2016; Durbin et al., 2017; Zahavich et al., 2017). Механистически, мутации NOTCH1 снижают способность связывать лиганд, нарушают S1 расщепление рецептора NOTCH в аппарате Golgi, и нарушают эпителиально-мезенхимный переход (Riley et al., 2011). Кроме того, мутации зародышевой линии в MIB1, который кодирует E3 ubiquitin ligase, способствующую эндоцитозу NOTCH лигандов, приводят к left ventricular non-compaction (LVNC) в аутосомно-доминантных родословных (Luxan et al., 2013). иокардиальные Mib1 мутации у мышей вызывают экспансию компактного миокарда, пролиферирующего в незрелые трабекулы и нарушающие развитие камер миокарда.
Проект encyclopedia of DNA elements (ENCODE) подтверждает, что более 80% геномной ДНК человека обладает биохимической функцией (Consortium, 2012). Большинство вызывающих болезнь вариантов, идентифицированных с помощью исследований геномных ассоциаций (GWAS), локализовано в некодирующих элементах ДНК, многие из которых внедрены в гиперчувствительных к DNase I (открытый хроматин) регионах (Maurano et al., 2012). GWAS при CHD привело к сходным находкам (Cordell et al., 2013; Hu et al., 2013; Mitchell et al., 2015; Hanchard et al., 2016). De novo варианты в энхансерных элементах были найдены при некоторых дефектах развития человека, включая CHD и нарушения нейрального развития (Short et al., 2018). Напр., варианты последовательностей в специфичном для конечностей энхансере ZRS, расположенном вблизи на расстоянии почти в 1 Mb от его гена мишени sonic hedgehog ( Shh), приводили к аномалиям конечностей, таким как преаксилярная полидактилия (Lettice et al., 2003). Варианты числа копий, затрагивающие топологические ассоциированные домены, также участвуют в нарушениях энхансеров и вызывают дефекты развития (Lupianez et al., 2015). Дистальные цис-регуляторные элементы были идентифицированы в TBX5, варианты которых ответственны за синдром Holt-Oram (Mcdermott et al., 2005; Smemo et al., 2012). Среди пациентов с синдромом Holt-Oram, три четверти имеют чаще всего CHD, с ASD и VSD. Гомозиготный вариант обнаружен в энхансере примерно на 90 kb ниже TBX5, он связан с изолированными ASD и VSD в когорте пациентов с несиндромальными CHD. Этот вариант в один нуклеотид нарушает активность энхансера по управлению экспрессии TBX5 в сердце трансгенных модельных мышей и рыбок данио (Smemo et al., 2012). Недавно WGS и chromatin immunoprecipitation секвенирование позволили исследователям увеличить количество генетических вариантов в некодирующих элементах ДНК, которые могли играть регуляторную роль в контроле транскрипции гена во время развития сердца (Zhao et al., 2017b; Richter et al., 2020). Некодирующие de novo variants (DNVs) достоверно увеличены у индивидов с CHD и потенциально способны осуществлять транскрипционные и пост-транскрипционные регуляторные эффекты на гены, критические для нормального морфогенеза сердца. Генетическая архитектура CHD в кардиальных регуляторных некодирующих DNVs будет в дальнейшем выяснена с помощью продвинутого WGS и технологий точного геномного редактирования.
Patient-Specific iPSCs for Modeling Genetics of CHD Хотя генетическая этиология идентифицирована примерно у 1/3 пациентов с CHD, экспериментальные модели с функциональной оценкой генетических вариантов, ассоциированных с CHD далеки от совершенства. Генетически преобразованные мыши были используются для изучения фундаментальной генетики развития сердца более 25 yearsлет. Мышиные модели способны воспроизводить некоторые аспекты развития сердца человека, благодаря сходству их ст. развития сердца и структуры сердца у взрослых (Majumdar et al., 2019). Однако, существуют существенные отличия в геномном содержании и физиологии у людей и мышей. Ортологические гетерозиготные варианты иногда не воспроизводят сходные проявления CHD при внесении в геном мышей. Происходящие от пациентов iPSCs, по-видимому, предоставляют уникальную платформу для изучения генетических механизмов CHDs, т.к. они содержат всю генетическую информацию исходных затронутых индивидов. В комбинации с CRISPR/Cas9 геномным редактированием, геномикой одиночных клеток и кардиальным органоидами, специфичные для пациентов iPSCs смогут существенно дополнять мышиные генетические модели CHD и иллюстрировать новые перспективы генетической этиологии CHD для будущих точной диагностики и лечения.
iPSCs человека являются многообещающими моделями для изучения генетических механизмов причин изолированных CHD, вызываемых дефектами одиночных генов. В дополнение к клеточно-автономным наследственным сердечным болезням, таким как синдром длинного QT (Moretti et al., 2010; Itzhaki et al., 2011), желудочковая тахикардия (Zhang et al., 2014; Sleiman et al., 2020) и дилятационная кардиомиопатия (Sun et al., 2012; Hinson et al., 2015), iPSCs пациентов были использованы, чтобы смоделировать некоторые типы CHD, включая BAV и calcific aortic valve disease (CAVD) (Theodoris et al., 2015), supravalvular aortic stenosis (SVAS) (Ge et al., 2012), cardiac septal defects (Ang et al., 2016), Barth syndrome (Wang et al., 2014), и HLHS (Hrstka et al., 2017; Yang et al., 2017; Miao et al., 2020; Table 2). Человеческие iPSCs могут быть дифференцированы в желательные типы сердечно-сосудистых клеток, подходящие для изучения разных CHD (Protze et al., 2019), хотя незрелость, происходящих из iPSC, кардиомиоцитов (iPSC-CMs) является препятствием для воспроизведения физиологического сценария в сердце (Karbassi et al., 2020; Zhao et al., 2020b). Протоколы правильной кардиальной дифференцировки были оптимизированы, чтобы генерировать специфические подтипы (предсердные, желудочковые и узелковые) кардиомиоциты дя точного моделирования болезни (Zhang et al., 2011; Lee et al., 2017; Protze et al., 2017; Ren et al., 2019; Liang et al., 2020; Zhao et al., 2020a).
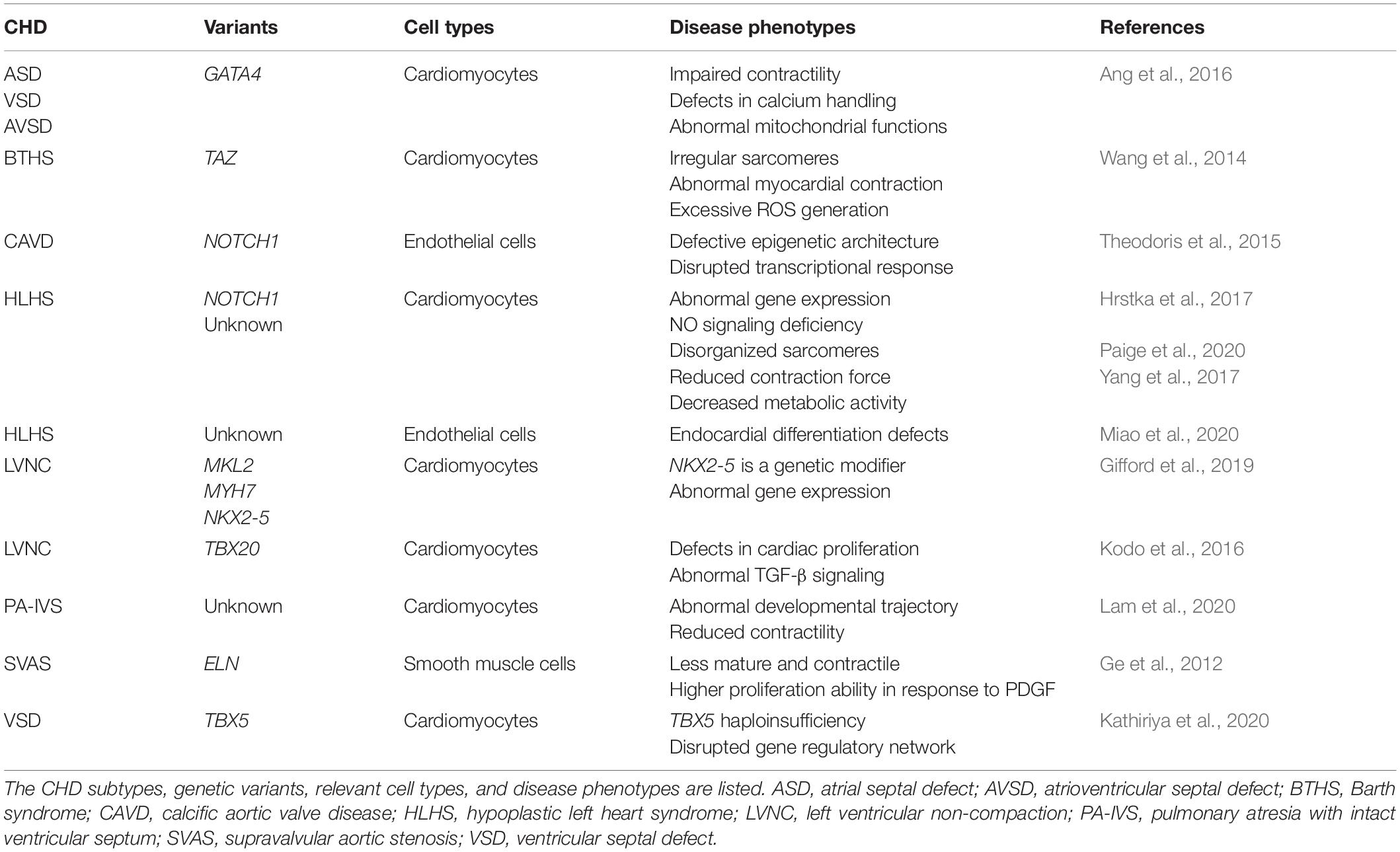 Table 2. Current iPSC models for studying disease mechanisms of CHD. Table 2. Current iPSC models for studying disease mechanisms of CHD. Человеческие iPSC модели CHD используют основные типы кардиальных клеток, такие как кардиомиоциты (CMs), сосудистые гладкомышечные клетки (SMCs) и эндотелиальные/эндокардиальные клетки (ECs), которые могут быть получены из специфических для пациента iPSCs для лаб. исследований. Эти, происходящие от пациента кардиальные клетки несут генетические варианты, позволяющие исследователям изучать механизмы болезни в чашках Петри (Table 2). Напр., патогенные GATA4 варианты вызывают дефекты кардиальных перегородок и кардиомиопатии. Гетерозиготные варианты GATA4 (G296S missense) сцеплены со 100% проявляющимися ASD, VSD, AVSD или PS (Garg et al., 2003). Человеческие iPSC-CMs от гетерозиготных GATA4-G296S пациентов обнаруживают нарушения сократимости, дефекты способности обслуживания кальция и аномалии митохондриальных функций (Ang et al., 2016). Молекулярный анализ выявил, что мутантный GATA нарушает рекрутирование TBX5, который соединяется с кардиальным супер-энхансером и приводит к нарушению регуляции генов, связанных с образованием кардиальных перегородок. В др. исследовании, Theodoris et al. (2015) получали iPSCs от пациентов с BAV и CAVD, которые были сцеплены с гаплонедостаточностью NOTCH1. В происходящих от iPSC эндотелиальных клетках (iPSC-ECs), NOTCH1 гетерозиготные нонсенс варианты нарушают эпигенетическую архитектуру связанных с NOTCH1 энхансеров и вызывают депрессию анти-остеогенных и противовоспалительных сетей регуляции генов в ответ на гемодинамический shear стресс (Theodoris et al., 2015). Более того, та же самая группа недавно использовала комбинацию технологии человеческих iPSC, компьютерного обучения и сетевой анализ для идентификации эффективного терапевтического кандидата XCT790 для предупреждения и лечения болезни аортальных клапанов у мышиной модели, продемонстрировав возможное фармакогенетическое применение специфичных для CHD пациентов iPSCs (Theodoris et al., 2020). Ge et al. (2012) использовали происходящие из iPSC гладкомышечные клетки (iPSC-SMCs) для изучения, как вариант гена elastin (ELN) приводит к сужению или блокдае восходящей аорты при SVAS. SVAS iPSC-SMCs, содержащие варианты ELN оказываются менее зрелыми и контрактильными и обнаруживают меньшие по размеру сети из пучков актиновых филамент из гладких мышц по сравнению с нормой. Такие SVAS iPSC-SMCs обнаруживают более высокую пролиферативную способность и величину миграции в ответ на platelet-derived growth factor (PDGF), демонстрируя, что SVAS iPSC-SMCs воспроизводят патологические свойства SVAS пациентов и могут предоставить новую информацию для будущей терапии. Человеческие iPSCs были использованы для изучения сложной генетики CHD вместе с трансгенными мышиными моделями и клинической генетикой. Недавнее исследование выявило, что варианты NKX2-5 выступают в качестве генетических модификаторов семейной LVNC кардиомиопатии, проявляющейся в разных возрастах (Gifford et al., 2019). Человеческие iPSCs были созданы, несущими наследственные компаундные гетерозиготные варианты в MKL2, MYH7 и NKX2-5 , тогда как созданные генетически преобразованные мыши также несли ортологические варианты. Путем анализа фенотипов из трансгенных сердец мышей и пациентов iPSC-CMs, были идентифицированы NKX2-5 варианты в качестве генетических модификатаоров для этой кардиомиопатии с олигогенным наследованием. В др. исследовании, линии LVNC iPSC были получены от пациентов с вариантами TBX20 (Kodo et al., 2016). LVNC iPSC-CMs обнаруживали дефекты пролиферации, которые были обусловлены аномальной активацией передачи сигналов TGF-β. У мышей с избыточная экспрессия TGF-β1 приводила к аресту развития сердца, нарушению экспансии эмбриональных кардиомиоцитов и соотношения трабекулярного и компактного слоя в левом желудочке. Недавно Kathiriya с колл. сгенерировали нокаутные по TBX5 человеческие iPSC линии с разными дозами (гетерозиготные и гомозиготные) и осуществили секвенирование РНК одиночных клеток и проанализировали регуляторные сети. TBX5 гаплонедостаточность меняла экспрессию связанных с CHD генов и пониженная доза гена TBX5 разрушала сети регуляторных генов в iPSC-CMs человека. Аномальные генетические взаимодействия между Tbx5 и Mef2c приводят к дефектам образования межжелудочковой перегородки у трансгенных мышей с редуцированной дозой гена Tbx5 (Kathiriya et al., 2020).

Figure 1. An integrated patient-specific iPSC model for studying genetics of CHD. Whole genome sequencing of CHD patients identifies prospective genetic variants that are retained in patient-specific iPSCs. Cardiovascular defects in CHD are recapitulated in patient iPSC-derived cardiac cells, such as cardiomyocytes (CMs), endocardial/endocardial cells (ECs), vascular smooth muscle cells (SMCs), and fibroblasts (FBs). Genetic variants associated with a CHD phenotype are corrected in diseased iPSCs and introduced to healthy iPSCs using CRISPR/Cas9 genome editing tools. These genome-edited iPSCs are further investigated to validate the cause-effect relationship between a given genetic variant and a CHD phenotype. In parallel, orthologous variants are genetically introduced into animal models (rodents, pigs, zebrafish, etc.) in order to investigate the in vivo effects of a given human variant on heart development. Together, an integrated model including both genome-edited human iPSCs and transgenic animals will yield a more comprehensive illustration of the genetic basis of CHD in the new era of genomic medicine. Синдром гипопластичного левого сердца является тяжелой формой CHD, характеризующейся атрезией аортальных и митрального клапанов или стенозом, приводящими к гипоплазии левого желудочка и аорты (Saraf et al., 2019). Благодаря HLHS имеется сильный генетический компонент, генетическая этиология HLHS сложна (Mcbride et al., 2005). Более того, мышиные модели неспособны полностью воспроизводить клинический фенотип (Liu et al., 2017; Grossfeld et al., 2019). Используя происходящие от пациентов с HLHS iPSC-CMs, многие исследования продемонстрировали патогенную связь NOTCH1 вариантов с HLHS (Theis et al., 2015; Durbin et al., 2017; Hrstka et al., 2017; Yang et al., 2017). HLHS iPSCs, несущие варианты NOTCH1, обнаруживают нарушения способности генерировать кардиальные предшественники, а HLHS iPSC-CMs обнаруживают дезорганизованные структуры саркомеров и саркоплазматического ретикулума, а также притупленную реакцию на лекарства (Yang et al., 2017). Др. независимое исследование подтвердило, что HLHS iPSCs обнаруживают дефицит в дифференцировке кардиомиоцитов и пути передачи сигналов NOTCH (Hrstka et al., 2017). Кроме того, аномалии пути nitric oxide (NO) обнаруживаются в спецификации кардиального клона HLHS iPSCs с NOTCH1 мутациями. Добавление малых молекул может восстанавливать кардиогенез, указывая на потенциальную терапевтическую мишень для пациентов с HLHS. Это исследование согласуется с врожденными кардиальными аномалиями, наблюдаемыми у Notch1+/-; Nos3-/- трансгенных мышей и демонстрирует, что взаимодействие между путями NO и передачи сигналов NOTCH необходимо для собственно развития левосторонних кардиальных структур, включая аортальный клапан (Bosse et al., 2013; Koenig et al., 2016). Недавно, Miao et al. (2020) подчеркнули вклад эндокардиальных дефектов в патогенез HLHS, используя iPSCs пациентов и секвенирование РНК одиночных клеток у плодов человека в ходе развития левого желудочка. Хотя генетические причины этих HLHS iPSCs неясны, эндокардиальные нарушения приводят к аномалиям эндотелиального-к-мезенхимному переходу, снижению пролиферации и созревания кардиомиоцитов и разрушению передачи сигналов fibronectin-integrin. Др. исследование Mikryukov et al. (2021) установило критическую роль BMP10 в спецификации и поддержании эндокардиальных клеток из iPSCs человека. Эти происходящие из iPSC эндокардиальные клетки могут индуцировать трабекуляцию из iPSC-CMs и генерировать подобные интерстициальным клетки клапанов, которые оказываются многообещающими in vitro моделях для изучения дефектов кардиальных клапанов и LVNC. Поскольку межклеточные коммуникации между эндокардом и миокардом являются важными для нормального развития желудочков (Macgrogan et al., 2018), то дальнейшие исследования смогут проиллюстрировать, как аномальные взаимодействия передач сигналов приводят к гипоплазии левого желудочка с использованием HLHS iPSC-CMs и iPSC-ECs.
Основной проблемой при изучении генетики HD отсутствие моделей по функциональной оценке генетических вариантов, которые обнаруживаются при массивном секвенировании генома. Хотя iPSC модели всё больше участвуют в исследованиях вклада генетических вариантов в развитие CHD, необходимо тщательно анализировать наиболее подходящие подходы. Человеческие iPSC-CMs являются подобными плодным кардимиоцитам и обнаруживают незрелые структурные и физиологические характеристики. Напр., iPSC-CMs не имеют зрелых структур из миофибрилл и T-трубочек и они неправильно располагаются по сравнению с палочковидными взрослыми кардиомиоцитами (Karbassi et al., 2020; Zhao et al., 2020b). Приложены огромные усилия для обеспечения структурной и функциональной зрелости iPSC-CMs, включая добавление тироидных и глюкокортикоидных гормонов (Parikh et al., 2017), физическое и электрическое кондиционирование (Ronaldson-Bouchard et al., 2018), и совместное культивирование со стромальными клетками в 3D кардиальных микро-тканях (Giacomelli et al., 2020). Кроме того, iPSC-CMs в основном культивируются как 2D структуры, которые отличаются от 3D структур человеческого сердца. Происходящие от iPSC кардиальные органоиды могут стать лучшими моделями, как предоставляющие 3D замену для человеческого сердца (Rossi et al., 2018; Richards et al., 2020). Однако, всё ещё не определено , действительно ли кардиальные органоиды могут воспроизводить сценарии возникновения CHD. Следовательно, любая базирующаяся на iPSC модель остается системой in vitro, которая фундаментально отлична от in vivo условий. Хотя животные модели лучше всего предоставляют in vivo условия, животные отличаются от человека по физиологии и геномике и не могут иметь клиническое значение. Поэтому мы предлагаем интегрированную модель, которая включает специфичные для пациента iPSCs с трансгенными животными (Figure 1). Мы полагаем, что генетические варианты, ассоциированные с CHD проявлениями, тестируемые с помощью отредактированных по геному iPSCs, происходящими от пациентов и клинически подходящие важны, тогда как ортологичные варианты будут генетически вноситься в животные модели (грызуны, свиньи, рыбки данио и т.д.), чтобы исследовать in vivo функции. Комбинирование человеческих iPSCs и трансгенных животных позволит нам более всеобъемлюще проиллюстрировать патогенетические механизмы CHD.
Outlook Недавние успехи CRISPR/Cas9 редактирования генома (Adli, 2018), геномики одиночных клеток (Tanay and Regev, 2017) и органоидов (Rossi et al., 2018) помогли открытию новых механизмов возникновения CHD, с использованием пациент- и болезнь-специфических iPSCs. Технологии точного редактирования геномов могут быть использованы для коррекции данного варианта iPSCs от пациента и дальнейшие исследования проявлений болезни могут спасать генетически откорректированные изогенные кардиальные клетки (Hockemeyer and Jaenisch, 2016; Deacon et al., 2019). Одновременно этот вариант может быть внесен в линию здоровых iPSC с новым генетическим фоном, чтобы протестировать , действительно ли это достаточная причина для проявлений болезни. Более того, олигогенное наследование CHD может быть исследовано на iPSCs от пациентов путем одновременной коррекции или внесения комбинации множественных генетических вариантов (Gifford et al., 2019). Анализ RNA-seq одиночных клеток в сердце человека и мыши предоставляет беспрецедентный источник траекторий кардиального развития in vivo с разрешением в одну клетку и это позволит наметить (blueprint), как детерминация нормальных клеточных судеб нарушается при генетических пертурбациях и патологических состояниях, таких как CHD (Cui et al., 2019; De Soysa et al., 2019; Litvinukova et al., 2020; Paik et al., 2020). Транскрипционное профилирование одиночных клеток iPSCs от здоровых и больных во время кардиальной дифференцировки позволит расшифровать, как данный генетический вариант влияет на кардиальную дифференцировку и онтогенетические траектории и раскрыть новую и молекулярную информацию патогенеза CHD (Churko et al., 2018; Kathiriya et al., 2020; Lam et al., 2020; Miao et al., 2020; Paige et al., 2020). Поскольку развитие сердца зависит от взаимодействия между многими типами клеток в эмбрионе, то кардиальные органоиды и 3D bio-printing могут послужить в качестве др. уровня моделирования болезни с использованием iPSCs пациента (Lee et al., 2019; Nugraha et al., 2019). Кардиальные органоиды содержат пространственную информацию о многих кардиальных типах клеток и базируются на 3-D платформе, чтобы изучать сложные взаимодействия между генотипами и фенотипами в нормальных и болезненных условиях с использованием пациент-специфичных iPSCs. Хотя кардиальные органоиды используются для моделирования вызываемой лекарствами токсичности и инфаркта миокарда (Richards et al., 2020), всё ещё затруднительно генерировать кардиальные структуры, такие как сердечные клапаны и перегородки, в которых часты дефекты при CHD с использованием современной технологии кардиальных органоидов.
|