Посещений: 
ВРОЖДЕННЫЙ ДИСКЕРАТОЗ
Нарушения биологии теломер
Understanding the evolving phenotype of vascular complications in telomere biology disorders Cecilia Higgs, Yanick J. Crow, Denise M. Adams et al.
 Angiogenesis
February 2019, Volume 22, Issue 1, pp 95–102|
| |
|
Нарушения биологии теломер (TBDs) проявляются в виде широкого круга сложных медицинских осложнений и вызываются патогенными вариантами генов биологи теломер зародышевой линии [1-4]. Эти нарушения известны также как синдромы коротких теломер [3]. Пациенты с dyskeratosis congenita (DC), имеют прототипическое TBD нарушение, которое обычно клинически проявляется в виде триады кружевная ретикулярная пигментация кожи, ротовая лейкоплакия и диспластичность ногтей. Он обусловливает высокий риск развития поражения костного мозга, легочный фиброз, бессосудистый некроз бедренных или плечевых костей, стеноз пищевода и др. мед. проблемы, включая высокий риск специфических раковых опухолей [5, 6]. Открытие патогенетических вариантов зародышевой линии в DKC1 (кодирующем dyskerin) в качестве причины X-сцепленного рецессивного DC, вперые показало, что аберрации в биологии теломер, приводящие к очень коротким теломерам могут быть причиной клинически различаемого фенотипического отклонения человека [7, 8]. Затем было обнаружено 13 дополнительных генов, ассоциированных с DC и родственными TBDs с de novo, аутосомно доминантным и аутосомно рецессивным наследованием (Fig. 1) [1, 9-11].
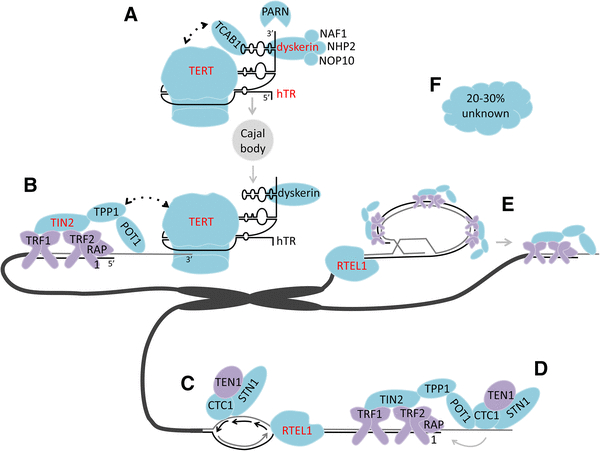 Fig. 1
Fig. 1
Factors and pathways impacted by mutations found in patients with TBDs. a Telomerase biogenesis and activation. b Telomerase recruitment. c Duplex telomeric DNA replication. d Terminal DNA end structure. e Resolution of t loop structure. fApproximate number of classic DC patients without detected mutation in one of the known genes. Subunits in blue shade and hTR have been found mutated in patients with DC. Those with red font are the most commonly mutated Клинический спектр TBDs расширяется по мере открытия новых генетических причин (Table 1). Некоторые пациенты, прежде всего те, что с преимущественно аутосомно доминантными болезнями, обусловленными мутациями TERT, TERC, PARN или RTEL1, могут развиваться как обладающие только одним признаком DC у взрослых в отсутствие mucocutaneous триады [1-3, 12-16]. Др. пациенты, включая тех, что имеют мутации в DKC1 , de novo TINF2 мутации или аутосомно рецессивные болезни могут обнаруживаться в раннем возрасте. Те, что с Hoyeraal-Hreidarsson синдромом, вариантом DC, обнаруживающие признаки иммунодефицита и гипоплазии мозжечка или синдрома Revesz, при котором признаки DC сочетаются с эксудативной ретинопатией и внутричерепными осложнениями [1, 6, 17], могут обнаруживаться у детей. Coats plus, редкое аутосомно рецессивное заболевание, обычно характеризуется внутричерепными кальцификациями, ассоциированными с кистами головного мозга, желудочно-кишечными (GI) кровотечениями, обусловленными telangiectasias, сосудистыми аномалиями сетчатки и аномальным заживлением костей [18, 19]. Coats plus оказывается сцепленным с TBD, если пациенты с DC имели также мутации в CTC1 [20, 21]. Сосудистые осложнения, такие как желудочно-кишечные кровотечения, telangiectatic аномалии, легочные артерио-венозные пороки развития, гепато-пульмональный синдром и аномалии сосудов сетчатки довольно часто встречаются у пациентов с TBDs. Выдвигается мнение о необходимости изучения этиологии и клинического ухода сосудистых осложнений при TBDs.
|