Посещений: 
РАЗЛИЧНЫЕ НАРУШЕНИЯ РЕГУЛЯЦИИ ТРАНСЛЯЦИИ
Разные причины болезней
Translation deregulation in human disease • Soroush Tahmasebi,
• Arkady Khoutorsky,
• Michael B. Mathews &
• Nahum Sonenberg
 Nature Reviews Molecular Cell Biologyvolume 19, pages791–807 (2018)
|
Advances in sequencing and high-throughput techniques have provided an unprecedented opportunity to interrogate human diseases on a genome-wide scale. The list of disease-causing mutations is expanding rapidly, and mutations affecting mRNA translation are no exception. Translation (protein synthesis) is one of the most complex processes in the cell. The orchestrated action of ribosomes, tRNAs and numerous translation factors decodes the information contained in mRNA into a polypeptide chain. The intricate nature of this process renders it susceptible to deregulation at multiple levels. In this Review, we summarize current evidence of translation deregulation in human diseases other than cancer. We discuss translation-related diseases on the basis of the molecular aberration that underpins their pathogenesis (including tRNA dysfunction, ribosomopathies, deregulation of the integrated stress response and deregulation of the mTOR pathway) and describe how deregulation of translation generates the phenotypic variability observed in these disorders.

|
Первое сообщение о связи мутации трансляционного компонента (митохондриальных tRNALys) с наследственным заболеванием человека (myoclonic epilepsy with ragged red fibres (MERRF) синдром) опубликовано в 1990 (ref.1). С тех порс список генетичеких забоеваний, вызываемых нарушением регуляции трансляции, увеличился значительно. Нарушения регуляции трансляции наблюдается при широком спектре болезней человека, включая иммунодефицит2,3, метаболичские нарушения4, нейрологические болезни5, а также при раке и во время вирусной инфекции6-10.
Трансляция является многоступенчатым процессом, представленным инициацией, элонгацией, завершением и рециклингом рибосом11. Во время канонической инициации цитозольные рибосомы рекрутируются на мРНК и сканируют их 5' untranslated region (5'UTR) на присутствие стартового кодана трансляции. При большинстве условий инициация является скорость-ограничивающей ступенью (происходит ~0.5-3.6 инициаций в мин. по сравнению со скоростью элонгации 3-10 аминокислот в сек.12-14), и поэтому она тонко регулируется. Несколько ключевых сигнальных путей, включая mammalian or mechanistic target of rapamycin (mTOR), mitogen activated protein kinases (MAPKs) и integrated stress response (ISR) пути, сходятся на ступени инициации, чтобы контролировать скорость синтеза белка в отве на разные внешние и внутренние сигналы15,16(Box 1). Контроль трансляции мРНК играет жизненно важную роль в регуляции экспрессии генов в эмбриональных и взрослых тканях. Поэтому дефекты процесса трансляции вредны для развития и физиологии организма. Митохондрии имеют параллельную систему трансляции, которая наиболее сходна с системой прокариот, чем с цитозольной трансляцией эукариот17 (Box 1), а мутации в этой системе вносят существенный вклад в болезни человека, нараушая аппарат генерации энергии клеток.
Мы подразделяем связаенные с трансляциией нарушения у людей на 4 группы: те, что участвуют в нарушении регуляции синтеза или функции tRNA, рибосомопатии, нарушения регуляции пути ISR и нарушения регуляции пути mTOR. Однако, имеется существенное перекрывание между этими категориями. Напр., с одной стороны, нарушения tRNA или функции митохондрий при разных заболеваниях запускают активацию ISR и подавляют путь mTOR18. С др стороны, транскрипционные и трансляционные мишени путей ISR и mTOR играют критические роли в бмогенезе tRNA, митохондрий и рибосом19.
Недавние успехи по секвенированию всего экзома и генома предоставли важные знания о новых мутациях, вызывающих болезни, которые затрагивают факторы трансляции 20-22, приводящие к более детальному пониманию патогенеза болезней и терапевтических возможностей. Мы впервые обсуждаем болезни человека, связанные с дефектами биогенеза митохондриальных tRNA, мутациями, затрагивающими цитозаольные tRNAs, aminoacyl-tRNA synthetases (ARSs; известены также как tRNA лигазы) и с факторами трансляционной элонгации (Figs 1,2; Supplementary table 1).
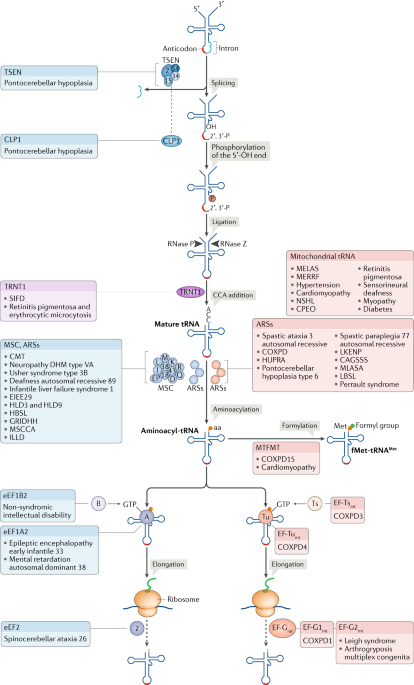 Fig. 1: Defects in tRNAs, aminoacyl-tRNA synthetases and translation elongation factors.
Fig. 1: Defects in tRNAs, aminoacyl-tRNA synthetases and translation elongation factors.
Diseases associated with cytosolic and mitochondrial defects are marked in blue and pink, respectively; diseases associated with both are marked in purple. tRNA splicing is affected by mutations in components of the tRNA-splicing endonuclease (TSEN) complex (TSEN2, TSEN15, TSEN34 and TSEN54) and in the polynucleotide kinase CLP1 (also part of TSEN). CCA tRNA nucleotidyltransferase 1, mitochondrial (TRNT1) catalyses CCA addition to tRNA 3?-ends. The tRNA multi-synthetase complex (MSC) comprises the aminoacyl-tRNA synthetases (ARSs) lysine-tRNA ligase (K), arginine-tRNA ligase, cytoplasmic (R), glutamine-tRNA ligase (Q), methionine-tRNA ligase, cytoplasmic (M), isoleucine-tRNA ligase, cytoplasmic (I), aspartate-tRNA ligase, cytoplasmic (D), leucine-tRNA ligase, cytoplasmic (L), bifunctional glutamate/proline-tRNA ligase (EP) and the scaffolding MSC auxiliary components p43, p38 and p18. Formylation of tRNAMet by mitochondrial methionyl-tRNA formyltransferase, mitochondrial (MTFMT) is required to generate the initiator tRNA fMet-tRNAMet. Elongation factor Tu, mitochondrial (EF-Tumt) and eukaryotic elongation factor 1-A2 (eEF1A2) deliver aminoacyl-tRNAs to the mitochondrial and cytosolic ribosome, respectively. EF-Tsmt and eEF1B2 are guanine nucleotide exchange factors for EF-Tumt and eEF1A, respectively. eEF2 is a GTPase and translocase that mediates ribosome movement on mRNA. There are two eEF2 homologues in mitochondria: EF-G1mt is a translocase, whereas EF-G2mt functions in ribosome recycling. CAGSSS, cataracts, growth hormone deficiency, sensory neuropathy, sensorineural hearing loss and skeletal dysplasia; CMT, Charcot-Marie-Tooth; COXPD, combined oxidative phosphorylation deficiency; CPEO, chronic progressive external ophthalmoplegia; DHM, distal hereditary motor; EIEE29, early infantile epileptic encephalopathy 29; GRIDHH, growth retardation, intellectual developmental disorder, hypotonia and hepatopathy; HBSL, hypomyelination with brainstem and spinal cord involvement and leg spasticity; HLD, hypomyelinating leukodystrophy; HUPRA, hyperuricaemia, pulmonary hypertension, renal failure in infancy and alkalosis; ILLD, interstitial lung and liver disease; LBSL, leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate; LKENP, leukoencephalopathy, progressive, with ovarian failure; MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes; MERRF, myoclonic epilepsy with ragged red fibres; MLASA, myopathy, lactic acidosis and sideroblastic anaemia; MSCCA, microcephaly, progressive, seizures, cerebral and cerebellar atrophy; NSHL, non-syndromic hearing loss; SIFD, sideroblastic anaemia with B cell immunodeficiency, periodic fevers and developmental delay.
Fig. 2: Human diseases linked to mitochondrial or cytosolic tRNA modifications.
All factors involved in tRNA modification are encoded in the nuclear genome. Diseases associated with cytosolic and mitochondrial defects are marked in blue and pink, respectively; diseases associated with both cytosolic and mitochondrial defects are marked in purple. ?, pseudouridine; ?m5s2U, 5-taurinomethyl-2-thiouridine; ?m5U, 5-taurinomethyluridine; ADAT3, probably inactive tRNA-specific adenosine deaminase-like protein 3; ALS, amyotrophic lateral sclerosis; CDKAL1, CDK5 regulatory subunit-associated protein 1-like 1; COXPD, combined oxidative phosphorylation deficiency; EKC, endopeptidase-like and kinase associated to transcribed chromatin; FTSJ1, protein ftsJ homologue 1; GTPBP3, GTP-binding protein 3; i6A, N6-(dimethylallyl)adenosine; KEOPS, kinase, endopeptidase and other proteins of small size; LAGE3, L antigen family member 3; m1G, 1-methylguanosine; m22G, N2,2?-O-dimethylguanosine; m5C, 5-methylcytidine; m7G, 7-methylguanosine; mcm5U, 5-methoxycarbonylmethyluridine; MLASA, myopathy, lactic acidosis and sideroblastic anaemia; ms2t6A, 2-methylthio-N6-threonylcarbamoyladenosine; MSSGM1, microcephaly, short stature and impaired glucose metabolism 1; MTO1, MTO1 homologue, mitochondrial; ncm5U, 5-carbamoylmethyluridine; Nm, 2?-O-methylnucleotides; NSUN, NOL1/NOP2/Sun domain family member; OSGEP, O-sialoglycoprotein endopeptidase; PRPK, p53-related protein kinase (also known as TP53RK); PUS, tRNA pseudouridylate synthase; t6A, threonylcarbamoyladenosine; TRMT1, tRNA (guanine(26)-N(2))-dimethyltransferase; TRMT5, tRNA (guanine(37)-N1)-methyltransferase; TRMT10A, tRNA methyltransferase 10 homologue A; TRMU, tRNA 5-methylaminomethyl-2-thiouridylate methyltransferase; TPRKB, TP53RK-binding protein; TRIT1, tRNA dimethylallyltransferase; WDR4, WD repeat-containing protein 4.
Fig. 3: A simplified overview of ribosome biogenesis.
a | The precursor to 18S, 5.8S and 28S ribosomal RNAs (rRNAs) is transcribed in the nucleolus by RNA polymerase I (Pol I), whereas 5S rRNA is transcribed in the nucleoplasm by Pol III. After processing and modification, the 18S, 5.8S and 28S rRNAs assemble with 5S rRNA and ribosomal proteins (RPs), which are synthesized in the cytoplasm and imported into the nucleolus to form pre-40S and pre-60S ribosomal subunits. Pre-40S and pre-60S subunits are then exported to the cytoplasm, where they undergo further maturation. b | Three models explaining tissue-specific phenotypes of ribosomopathies are depicted. The ribosome concentration model proposes that ribosome dysfunction affects global translation but that certain mRNAs and cell types are more sensitive to the change in ribosomal function. By contrast, the specialized ribosome model proposes that the composition of ribosomes varies depending on the tissue and stress conditions and that this unique composition determines which subset of mRNAs is translated. The tumour suppressor p53-mediated model suggests that impaired ribosome biogenesis activates the p53 pathway to induce cell cycle arrest or apoptosis in affected cell types. CHH, cartilage hair hypoplasia; DBA, Diamond-Blackfan anaemia; MDM2, E3 ubiquitin-protein ligase; SDS, Schwachman-Diamond syndrome.
Fig. 4: Integrated stress response-related diseases.
Cellular stress leads to activation of the integrated stress response through phosphorylation of the ? subunit of eukaryotic translation initiation factor 2 (eIF2?). This is mediated through stimulation of one of four eIF2? kinases: general control nonderepressible 2 (GCN2), PKR-like endoplasmic reticulum (ER) kinase (PERK), protein kinase RNA-activated (PKR) and haem-regulated inhibitor (HRI), each of which is responsive to different cellular stressors (not shown). Growth arrest and DNA damage-induced protein GADD34 (also known as PPP1R15A) and constitutive reverter of eIF2? phosphorylation CReP (also known as PPP1R15B) are regulatory subunits of PP1, which can dephosphorylate eIF2?. Phosphorylation of eIF2? attenuates general translation by inhibiting the assembly of the eIF2-GTP-Met-tRNAiMet ternary complex, but also stimulates translation of stress response mRNAs such as those encoding activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP), which have upstream open reading frames in their 5? untranslated regions and thus escape the inhibition of general translation by an indirect mechanism. eIF2B is a multisubunit (comprising eIF2B?, eIF2B?, eIF2B?, eIF2B? and eIF2B?) guanine nucleotide exchange factor for eIF2. DNAJC3, DnaJ homologue subfamily C member 3; GCN1, general control of amino-acid synthesis 1-like protein 1; IER3IP1, immediate early response 3-interacting protein 1; MRXSBRK, mental retardation, X-linked, syndromic, Borck type; PVOD, pulmonary veno-occlusive disease; SIL1, nucleotide exchange factor SIL1; VWM, vanishing white matter; WRS, Wolcott-Rallison syndrome.
Full size image
Fig. 5: Human diseases linked to the mTOR complex 1 pathway.
Diseases linked to growth factor-dependent activation of mTOR complex 1 (mTORC1) are highlighted in blue, and diseases linked to amino acid-dependent mTORC1 activation are in green. Brown denotes the diseases that are shared by both pathways. In response to growth factors and insulin, the PI3K-AKT pathway stimulates mTORC1 through inhibition of the tuberous sclerosis complex (TSC), a negative regulator of the small GTPase GTP-binding protein RHEB. Amino acids activate mTORC1 through activating RAS-related GTP-binding protein (RAG) GTPases. Both growth factor-dependent and amino acid-dependent pathways must be activated to stimulate mTORC1 activity. mTORC1 controls translation initiation through phosphorylation of several effectors. In addition to translation initiation, mTORC1 controls elongation through phosphorylation of ribosomal protein S6 kinases (S6Ks) and eukaryotic elongation factor 2 (eEF2) kinase (eEF2K). The activity of RNA polymerase I (Pol I) and Pol III and the transcription of mRNAs encoding ribosomal proteins are also controlled by mTORC1 through S6Ks and repressor of Pol III transcription MAF1 homologue (MAF1). 4E-BPs, eIF4E-binding proteins; ARCL2A, cutis laxa, autosomal recessive, type IIA; ATP6AP1, vacuolar proton pump subunit S1; ATP6V0, vacuolar proton translocating ATPase 116?kDa subunit; ATP6V1B1, vacuolar proton pump subunit B1; BRRS, Bannayan-Riley-Ruvalcaba syndrome; cyclin D2, G1/S-specific cyclin D2; CLOVE, congenital lipomatous overgrowth, vascular malformations and epidermal nevi; DEPDC5, DEP domain-containing 5; eIF, eukaryotic translation initiation factor; FCORD2, focal cortical dysplasia type II; FFEVF, familial focal epilepsy with variable foci; FLCN, folliculin; GATOR1, GAP activity towards Rag 1 ; HIHGHH, hypoinsulinaemic hypoglycaemia with hemihypertrophy; IGF1R, insulin-like growth factor 1 receptor; INSR, insulin receptor; IRS, insulin receptor substrate; ITFG2, integrin-? FG-GAP repeat-containing protein 2; KICSTOR, KPTN-ITFG2-C12ORF66-SZT2; KPTN, kaptin; LARP1, La-related protein 1; MAPBPIP (p14), (mitogen-activated protein-binding protein)-interacting protein; MCAP, megalencephaly-capillary malformation-polymicrogyria; MPPH, megalencephaly-polymicrogyria-polydactyly-hydrocephalus; NIDDM, non-insulin-dependent diabetes mellitus; p110, PI3K catalytic subunit; p85, PI3K regulatory subunit; PDCD4, programmed cell death protein 4; SHORT, short stature, hyperextensibility, hernia, ocular depression, Rieger anomaly and teething delay; SZT2, seizure threshold 2 protein homologue; TBC1D7, TBC1 domain family member 7; TSC1, hamartin; TSC2, tuberin; v-ATPase, lysosomal vacuolar H+-ATPase.
|
Box 1 mRNA translation in the cytosol and in mitochondria
Nuclear-encoded eukaryotic mRNAs undergo several steps of processing in the nucleus, which include the addition of a 5?-terminal cap (7-methylguanosine (m7G)) at the 5?-end and a poly(A) tail at the 3?-end, followed by internal base methylation, splicing and export to the cytosol (see the figure, part a). Ribosomes are recruited to the mRNA through the coordinated activity of multiple translation initiation factors. Two protein complexes, eukaryotic translation initiation factor 4F (eIF4F), which comprises eIF4E (a cap-binding protein), eIF4G (a scaffolding protein) and eIF4A (an RNA helicase), and the ternary complex, which comprises eIF2, GTP and the initiator tRNA (Met-tRNAiMet), have key roles in translation initiation. mRNA circularization occurs through the interaction of eIF4G with poly(A)-binding protein (PABP). The eIF4F complex unfolds secondary structures in the 5? untranslated region (5?UTR) of mRNAs for translation initiation. In addition, an interaction (not shown) between eIF4G and eIF3 brings the 40S small ribosomal subunit, as a component of the 43S preinitiation complex, into the vicinity of the mRNA 5?-end to start the scanning process. The integrated stress response (ISR) and mTOR complex 1 (mTORC1) control translation initiation through regulation of the ternary complex and the eIF4F complex, respectively. Interaction of eIF4B with eIF4A increases the helicase activity of eIF4A. Unlike nuclear mRNAs, mitochondrial mRNAs (mt-mRNAs) are uncapped and have a short (up to three nucleotides in length) or no 5?UTR, obviating the need for initiation factors to unwind the 5?UTR (see the figure, part b).
AUG, translation initiation codon; mtDNA, mitochondrial DNA; mt-rRNA, mitochondrial ribosomal RNA; mt-tRNA, mitochondrial tRNA; OXPHOS, oxidative phosphorylation complexes; STOP, stop codon.
|
Deregulation of tRNA function
Компоненты аппарата трансляции в митохондриях - митохондриальные tRNAs (mt-tRNAs), tRNA модифицирующие энзимы, ARSs, факторы элонгации и рибосомальные белки - часто мутантные при митохондриальных болезнях (Supplementary table 1). Митохондриальные болезни встрачаются среди наиболее часто наследемых нарушений у человека. Они результат мутаций ядерных или митохондриальных генов. Болезни, ассоциированнные с ядерными генами, обычно аутосомно рецессивные, проявляются в раннем периоде жизни и обнаруживают мультисистемые фенотипические отклонения с фатальными последствиями. Напротив, болезни, вызываемые мутациями в генах митохондриальной ДНК (mtDNA), наследуются от матери и часто менее тяжелые. Большинство mt-tRNAs кодируются одиночным геном, тогда как ~50 цитозольных выидос tRNA кодируются ~500 ядерными генами 23. Это может объяснить, почему неизвестны болезни человека, вызываемые мутациями кодируемых в ядре tRNAs. Тем не менее, мутации сплайсинга tRNA и модифицирующих факторов были идентифицрованы при некоторых нарушениях у человек, прежде всего при нейродегенеративных болезнях 24-27.
Mitochondrial tRNA
Митохондриальный геном, передающийся исключительно через женскую зародышевую линию, кодирует37 генов. включая 22 mt-tRNAs и 2 рибосомальные РНК (rRNAs) (16S и 12S). Оставшиес 13 генов кодируют белки, которые участвуют в oxidative phosphorylation (OXPHOS). Большинство митохондриальных белков (включая те, что участвуют в митохондриальной трансляции) кодируется в ядре, транслируется с мРНК в цитозоле и транспортируется в митохондрии. Митохондриальные гены подвергаются значительно большему числу вредных мутаций, чем ядерные гены28. Одностороннняя материнская передача, отсутствие интронов, повторяющиеся элементы и рекомбинация, а также низкий уровень точности репликации mtDNA полимеразы вносят вклад в высокий уровень мутаций в митохондриальных генах29,30. К счастью, вредные дефекты мутаций ослабляются существованием нескольких копий (2-10) митохондриального генома в каждой митохондрии и многочисленность митохондрий в каждой клетке, так что болезнь проявляется только, когда количество неправильно функционирующих митохондрий превышает допустимый порог31. Нарушения функции mt-tRNAs возникают в результате мутаций в mt-tRNA последовательностях или в результате дефектов в кодируемых в ядре энзимов, модифицирующих tRNA (Figs 1,2; Supplementary table 1). Принимая во внимание ограниченное число mt-tRNAs (22 mt-tRNAs кодируют 60 смысловых кодона митохондриального генетического кода), модификации mt-tRNA, особенно в шаткой (wobble) позиции (tRNA нуклеотид 34, который является первым (5') основанием антикодона), выполняют критическую роль в расширенном распозновании кодонов. Т.о., в контексте болезни мутации энзимов, модифицирующих tRNA, ограничивают декодирующую способность mt-tRNA. Дефект в системе OXPHOS является широко распространенным исходм нарушения регуляции трансляции в митохондриях32. Головной мозг и мышцы особенно чувствительны к активности OXPHOS из-за её высокой потребности в энергии. Однако, в большинстве случаев вызывающие болезнь мутации приводят к дефектам в органах иных, чем мышцы или нервная система, указывая на существование ген-специфиеских эффектов32.
Мутации в генах mt-tRNA имеют или мультиорганное или ткане-специфическое проявление (Fig. 1; Supplementary table 1). Два наиболее охарактеризованных мультиорганных синдрома ассоциированые с мутациями в mt-tRNA, это MERRF и mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS). MERRF в основном сцеплен с дефицитом митохондриального комплекса IV33, тогда как MELAS связывают преимущественно с дефектами митохондриального комплекса I. Мутации в некоторых mt-tRNA генах вызывают синдром cause MELAS (Supplementary table 1), хотя в большинстве случаев он вызывается мутациями митохондриями кодируемой tRNA leucine 1 (MT-TL1). Наиболее распространена мутация, расположенная в mt-tRNALeu wobble позиции (A3243G) и она вмешивается в 5-taurinomethyluridine (τm5U) модификации в этой позиции34 (Fig. 2). MERRF синдром может также возникать в резульате отсутствия модификации taurine в шаткой позиции mt-tRNALys). Очевидно, что отсутствие модификации taurine в mt-tRNALys мешает декодированию обоих кодонов Lys, тогда как отсутствие модификаций taurine в mt-tRNALeu влияет на декодирование только Leu кодона (UUG , но не UUA)35. Такая склонность к декодированию преимущественно затрагивает митохондриальный комплекс I, благодаря дефекту в трансляции митохондриями кодируемой мРНК NADH dehydrogenase 6 (MT-ND6), которая имеет высокое содержание UUG. Это также объясняет фенотипические отличия между MERRF и MELAS синдромами. В самом деле, пациенты, несущие точечную мутацию в MT-ND6, обнаруживают существенный дефект в активности комплекса I в мышцах и обнаруживают фенотип, сходный с синромом MELAS36.
Важность модификаций tRNA в шаткой позиции была недооценена при болезнях, вызываемых мутациями в энзимах, которые катализируют эти модификации (напр., в protein MTO1 homologue, mitochondrial, GTP-binding protein 3 (GTPBP3) и tRNA 5-methylaminomethyl-2-thiouridylate methyltransferase (TRMU; известой также как mitochondrial tRNA-specific 2-thiouridylase 1)) 37-39 (Fig. 2; Supplementary table 1). Модификации mt-tRNAMet особенно важны, т.к. они позволяют одиночной tRNA служить в качестве элонгатора tRNA (Met-tRNAMet) и инициатора tRNA (fMet-tRNAMet), чего не происходит в случае цитозоля, митохондрий прокариот или дрожжей. Methionyl-tRNA formyltransferase, mitochondrial (MTFMT) является ферментом, который модифицирует Met-tRNAMet в fMet-tRNAMet. Мутации в MTFMT оказались сцеплены с синдромом Leigh syndrome и cardiomyopathy 40 (Fig. 1). Кардиомиопатия, несиндромальная потеря слуха и внешняя офтальмоплегия являются примерами ткане-специфической патологии, вызываемой мутациями в mt-tRNA (Supplementary table 1). Мутации в некоторых генах mt-tRNA недавно были идентифицированы у пациентов с матерински наследуемой гипертензией 41, но молекулярные механизмы их действия остаются неизвестными.
Cytosolic tRNA
Шаткая позиция в цитозольных tRNAs также является предметом модификаций. Высоко консервативный гексамерный Elongator complex добавляет 5-methoxycarbonylmethyl (mcm5) и 5-carbamoylmethyl (ncm5) к уридиновому остатку в wobble позиции нескольких цитозольных tRNAs42(Fig. 2). Мутации потери функции Elongator complex protein 1 (ELP1; каркачный белок для Elongator) были идентифицированы у евреев Ashkenazi с семейной dysautonomia43,44,аутосомно рецессивной нейродегенеративной болезнью, затрагивающей сенсорные и аутономные нейроны. Мутация одиночного нуклеотида в интроне 20 донорского сайта связывания, была идентифицирована у более 99.5% индивидов с семейной dysautonomia44. Мутация собcтвенно вмешивается в сплайсинг мРНК ELP1, вызывая пропуск экзона 20. Интересно, что пропуск экзона 20 происходит более часто в нейронах, чем в остальных клетках, объясняя преимущественно нейродегенеративный фенотип при семейной dysautonomia и открывая возможность модификации сплайсинга для терапии эой болезни (напр., с помощью kinetin)45. Безусловно, мутации др. компоентов Elongator комплекса также оказались связаны с нейродегенеративными нарушениями46,47.
Нуклеотид 37 в tRNAs (первый нуклеотид, стоящий ниже антикодона) также является горячей точкой модификаций. Сильно законсервированный пентамерный KEOPS-EKC (kinase, endopeptidase and other proteins of small size (KEOPS)-endopeptidase-like and kinase associated to transcribed chromatin (EKC)) комплекс обеспечивает threonylcarbamoyladenosine (t6A) модификацию в этом положении, чтобы контролировать аккуратность и эффективность трансляции (Fig. 2). Недавнее исследование идентифицировало рецессивную мутацию в 4-й субъединице KEOPS-EKC комплекса у пациентов с почечено-нейрологическим забоеванием, известным как Galloway-Mowat синдром20,48. Делеция ортологических генов у мышей и рыбок данио воспроизводят некоторые из фенотипов20.
Нарушение регуляции функции tRNA также ассоциирует с мутациями, затрагивающими факторы сплайсинга tRNA. Pontocerebellar hypoplasia (PCH) является спектром из нейродегенеративных нарушений с ранним началом головного мозга, которые вызывают микроцефалию, судороги и умственную отсталость. Мутации в разных генах могут вызывать PCH, и несколько линий доказательств подтверждает, что в основе этого нарушения лежат дефекты глобального синтеза белка 49. Среди наиболее охарактеризованных этиологий PCH являются мутации, затрагивающие tRNA-splicing endonuclease (TSEN) комплекс (Fig. 1). Хотя только 6% tRNA генов человека содержат интроны, рецессивные мутации всех 4-х субъединиц комплекса TSEN оказались связанными с разными формами PCH 24,25. Удаление и лигация интронов tRNA нуждаются в активности cleavage and polyadenylation factor CLP1. CLP1, который был первой RNA киназой, идентифицированной у млекопитающих 50, взаимодействует с TSEN комплексом, чтобы способствовать сплайсингу tRNA 51. Недавно описаны гомозиготные мутации CLP1 у индивидов с PCH с проявлениями как в ЦНС, так и ПНС (PCH тип 10) 26,27.
Aminoacyl-tRNA synthetases
ARSs являются энзимами, катализирующими прикрепление аминокислот к соотв. tRNAs: 17 ARSs действуют в цитозоле, 18 функционируют в митохондриях и 2 действуют и в цитозоле и в митохондриях (glycine-tRNA ligase (GARS) и lysine-tRNA ligase (KARS)). Бифункциональные glutamate/proline-tRNA ligase (EPRS) осуществляет tRNA aminoacylation как Glu, так и Pro в цитозоле.
Mitochondrial aminoacyl-tRNA synthetases
Мутации митохондриальных ARSs мешают митохондриальным респираторным цепочкам и вызывают мультисистемные нарушения. Фенотипы этих мутаций варьируют от энцефалопатии до кардиомиопатии или sideroblastic анемии (Supplementary table 1). Эти редкие болезни являются аутосомно рецессивными и фатальными в первые годы жизни 52,53.
Cytosolic aminoacyl-tRNA synthetases
tRNA multi-synthetase complex (MSC), который осуществляет процесс aminoacylation, представлен 8 цитозольными ARSs и тремя каркасными белками (MSC auxiliary component p43 (известен также как AIMP1), MSC auxiliary component p38 (известен также как AIMP2) и MSC auxiliary component p18 (известен также как eEF1E1)) 54. Большинство мутаций цитозольных ARSs, вызывающих болезнь, щядят ЦНС (за немногими исключениями 55) (Supplementary table 1) и вызывают периферические нейропатии или болезнь Charcot-Marie-Tooth (CMT) (Fig. 1). Существуют специальные случаи мутаций гаплонедостаточности, которые дают исключительно периферические нейропатии или наследственные дистальные моторные нейропатии 56. CMT является наиболее распространенной наследственной полинейропатией (с показателем 1 на 2500 индивидов в США). Это генетически гетерогенное заболевание, которое затрагивает как сенсорные, так и двигательные нейроны посредством дегенерации аксонов или демиелинирования нейронов. Кстати, мутации 6 ARSs (alanine-tRNA ligase, cytoplasmic (AARS), GARS, histidine-tRNA ligase, cytoplasmic (HARS), KARS, methionine-tRNA ligase, cytoplasmic (MARS) и tyrosine-tRNA ligase, cytoplasmic (YARS)) сцеплены с болезнью CMT. Т.к. нервные окончания периферических нейронов удалены от тела нейрона, то локальная трансляция мРНК играет центральную роль в регуляции протома аксона. Это свойство, как полагают, делает периферические нейроны склонными к нарушениям цитозольных ARSs 57. Примечательно, что потеря активности aminoacylation не является основным последствием всех мутаций ARS, указывая, что неканонические функции ARSs играют роль в патогенезенекоторых связанных с ARS нарушениях 58. Некоторые мутации вызывают конформационные изменения в ARSs, которые способствуют взаимодействию энзимов с новыми партнерами и приводят к новым функциям 59,60. Существуют доказательства неканонический функций ARSs в ангиогенезе 61, иммунных реакциях 62и реакциях на повреждения ДНК 63.
Translation elongation factors
Мутации трех факторов элонгации (elongation factor Tu, mitochondrial (EF-Tumt), elongation factor Ts, mitochondrial (EF-Tsmt) и elongation factor G, mitochondrial (EF-Gmt)) и их цитозольные аналоги (eukaryotic translation elongation factor 1A (eEF1A), eEF1B и eEF2, соотв.) сцеплены с болезнями человека, которые в основном затрагивают ЦНС (Fig. 1; Supplementary table 1).
Ribosomopathies
Дефекты биогенеза и функции рибосом вызывают спектр болезней, наз. рибосомопатиями. Цитозольные рибосомы млекопитающих состоят из 4-х rRNAs и большого числа рибосомальных белков (имеется 80 стержневых рибосомальных белков). Сборка pre-40S и pre-60S частиц рибосом начинается в ядрышке и заканчивается в цитозоле (Fig. 3a). Болезни, ассоциированные с гаплонедостаточностью рибосомальных генов, неожиданно обнаруживают ткане-специфичные и иногда клеточно-специфичные фенотипы, при этом большинство нарушений приходится на клеточные клоны, происходящие из костного мозга и на скелетные или черепно-лицевые аномалии64,65 (Fig. 3a; Supplementary table 1).
Одной из первых описанных рибосомопатий стала Diamond-Blackfan anaemia (DBA), которая характеризуется драматическим снижением эритроидных предшественников в костном мозге, что сопровождается макроцитарной анемией и reticulocytopenia66. Эта врожденная анемия чаще всего диагеностируется во время первого года жизни. Свыше 50% пациентов с DBA имеют короткий рост, черепно-лицевые дефекты (расщепление губы или нёба), аномалии большого пальца рук (из трех фаланг) и врожденные пороки сердца. Мутации в гене, кодирующем 40S рибосомальный белок S19 (RPS19) отвечают за ~25% индивидов с DBA, тогда как мутации в др. рибосомальных белках менее распространены67(Supplementary table 1). DBA вызываются снижением биогенеза рибосом и нарушениями синтеза белка, но механизм предпочтительного проявления в эритроидных клетках костного мозга неясен68(Fig. 3a). Недавние исследования установили важность GATA-binding factor 1 (GATA1), который является критическим для регуляции транскрипции при эритропоэзе, в патогенезе DBA и предоставляют потенциальное объяснение эритроидной специфичности этого фенотипа69,70. Альтернативный сплайсинг мРНК GATA1 человека генерирует две изоформы GATA1, длинная изоформа, содержащая второй экзон, и короткая изоформа, лишенная этого экзона. Мутации в GATA1, которые снижат продукцию белка полной длины (путем вмешательства в сплайсинг или трансляцию длинной изоформы GATA1) могут вызывать DBA69,70. Кроме того, трансляция GATA1 мРНК особенно чувствительна к подавлению рибосомальных белков, это связано с более высоким порогом потребностей для инициации трансляции мРНК GATA169.
Хорошо изученный синдром, обладающий эритроидными дефектами, - 5q- синдром, вызываемый гетерозиготной делецией длинного плеча хромосомы 5 (del(5q)). Макроцитарная анемия, характеризующая это нарушение, вызывается гаплонедостаточностью гена RPS1471,72, тогда как гаплонедостаочность по др. генам в делетированном хромосомном регионе вносит вклад в не-эритроидный фенотип: так, гаплонедостаточность microRNAs miR-145 и miR-146a связаны с тромбоцитозом73, а гаплонедостаточность early growth response protein 1 (EGR1) сцеплена с клонаьным доминированием74. Недавнее исследование продемонстрировало важность La-related protein 1 (LARP1), являющегося регулятором трансляции и эффектором mTOR complex 1 (mTORC1)75, в патогенезе 5q- синдрома76. Анемия при DBA и 5q- синдроме ослабляется у животных моделей и пациентов при лечении с помощью L-leucine77,78. L-leucine-вызываемая активация mTORC1, способствует трансляции мРНК и биогенезу рибосом, как полагают, делает более благоприятными эти эффекты77. Растут доказательства возможной роли активторов mTORC1 в возникновении рака79, эти находки открывают терапевтические возможности для пациентов с DBA или 5q- синдромом и составляют базу для тестирования эффективности L-leucine при др. рибосомопатиях.
Др. синдромы, ассоциированные с мутациями в белках, участвующих в биогенезе рибосом, обнаруживают широкий круг симптомов. Schwachman-Diamond syndrome (SDS) характеризуется нарушениями функции экзокринной части поджелудочной железы, нарушениями гематопоэза и нейтропенией и дефектами костей80. SDS вызывается мутацией в гене, кодирующем ьеорк сощзревания рибосом mutation in the gene encoding ribosome maturation SBDS, который соединяется с 60S рибосомальной субъединицей и участвует в биогенезе 60S рибосом и процессинге РНК81 (Fig. 3a). Cartilage hair hypoplasia (CHH) является нарушением, характеризующимся карликовостью с короткими конечностями и тонкими, редкими волосами, а также дефектами клеточного иммунитета, гипопластической анемией и дисплазией нейронов кишечника82-84. Эта болезнь вызывается мутациями в гене, кодирующем RNA component of mtRNA processing endoribonuclease (RMRP), которая кодирует non-coding RNA компонент RNase mtRNA processing (MRP) комплекса, необходимого для процессинга rRNA82,85. Нарушения процессинга rRNA, участвующего в созревании 40S субъединицы, таже связаны с Bowen-Conradi синдромом и с несиндромальной Aplasia cutis congenita86,87. В то время как Aplasia cutis congenita является довольно легким заболеванием, которое чаще всего проявляется как дефект кожи скальпа, синдром Bowen-Conradi является фатальным врожденным заболеванием с многочисленными онтогенетическими дефектами и ранней смертью. Синдром Treacher Collins характеризуется дефектами роста без гематологических аномалий. Болезнь вызывается мутациями в RNA polymerase I и III subunit D (POLR1D) и RNA polymerase I and III subunit C (POLR1C), каждая из которых кодирует субъединицу как RNA polymerase I (Pol I) комплекса, так и Pol III комплекса, и в treacle ribosome biogenesis factor 1 (TCOF1); эти три кодируемых белка участвуют в транскрипции генов, кодирующих рибосомальную РНК, tRNA и др. малые РНК, и в метилировании rRNA88-90. Дефекты модификаций rRNA в целом могут быть ответственны за рибосомопатии. Напр., синдром X-linked dyskeratosis congenita, который неточно классифицируется как 'premature ageing syndrome', вызывается мутацией гена, кодирующего dyskerin (DKC1), который обеспечивает pseudouridylation rRNA91.
Механизмы, с помощью которых аберрантный биогенез рибосом затрагивает клеточные функции и является причиной изменчивости клинических проявлений рибосомопатий, остаются неизвестны 92,93. Модель концентрации рибосом предполагает, что нарушения регуляции трансляции специфических мРНК или снижение уровней глобальной трансляции могут быть связаны с рибосомальными мутациями, нарушающими клеточную активность 68 (Fig. 3b). Тканевая специфиченость рибосомопатий приписывается разной чувствительности трансляции специфических мРНК к дисфункции рибосом в разных тканях 69. Альтернативно, рибосомопатии могут возникать в результате дисбаланса в соотношениях рибосомальных белков, которые, как было установлено, активируют сигнальный путь tumour suppressor p53. Согласно этой модели, снижение синтеза определенного рибосомального белка ведет к нарушению биогенеза рибосом и накоплению несобранных рибосомальных белков. Эти белки соединяются с E3 ubiquitin-protein ligase MDM2 и их активность супрессируется 94,95. MDM2 негативно регулирует p53 путем целенаправленного воздействия на его деградацию протеосомами 96. Поэтому снижение активности MDM2 приводит к стабилизации и активации p53 (refs95,97-99). В свою очередь, это способствует аресту клеточного цикла и апоптозу, это непропорционально воздействует на быстро делящиеся гематопоэтические клетки. Инактивация p53 у мышей, моделирующих DBA (мутация в Rps19) 100 и 5q-синдром 101 устраняет некоторые фенотипические отклонения, включая гипоплазию эритроцитов, подтверждая тем самым мнение, что активация p53 вносит вклад в эритроидный дефицит при этих рибосомопатиях 102,103. Альтернативная гипотеза предполагает, что 'специализированные рибосомы' с уникальным составом рибосомальных белков и тканевым распределением необходимы для трансляции определенных мРНК 104,105. Потеря таких специализированных рибосом может объяснить тканевую специфичность дефицита рибосомальных белков (Fig. 3b). Роль p53-зависимого и p53-независимого путей в ответ на нарушения биогенеза рибосом и механизмы, лежащие в основе тканевой специфичности, является активной областью исследований 68,107.
The integrated stress response
ISR воспринимают разнообразные клеточные стрессы и опосредуют изменения в экспрессии генов, чтобы адаптироваться к стрессам. Разные стрессоры активируют 4 киназы, которые сходятся на одном сайте фосфорилирования: Ser51 (у людей) из α-субъединицы (or subunit 1) человеческого eukaryotic translation initiation factor 2 (eIF2α; известен также как eIF2S1). 4 киназы являются haem-regulated inhibitor (HRI; известен также как eIF2AK1), protein kinase RNA-activated (PKR; известен также как eIF2AK2), PKR-like endoplasmic reticulum (ER) kinase (PERK; известна также как eIF2AK3) и general control nonderepressible 2 (GCN2; известен также как eIF2AK4) (Fig. 4). Комплекс eIF2 участвует в инициации трансляции путем формирования третичного комплекса с инициатором Met-tRNA и GTP, которые доставляются в малую рибосомальную субъединицу (40S), чтобы сформировать 43S преиниационный комплекс (Box 1). Фосфорилирование eIF2α блокирует активность guanine nucleotide exchange factor eIF2B, который превращает GDP-связанный eIF2 в GTP-связанный eIF2. Фосфорилирование eIF2α снижает общую трансляцию, но избирательно повышает трансляцию мРНК, обладающих вышестоящими открытыми рамками считывания в своих 5'UTR, таких как те, что кодируют факторы транскрипции activating transcription factor 4 (ATF4) и C/EBP homologous protein (CHOP; известен также как DDIT3). Жизненно важная роль ISR в клеточной функции демонстрируется некоторыми болезнями, вызываемыми мутациями в генах этого пути, включая те, что кодируют eIF2α kinases PERK и GCN2, eIF2B, γ-subunit (subunit 3) в eIF2 (eIF2γ; известна также как eIF2S3) and protein phosphatase 1 regulatory subunit 15B (PPP1R15B) (Fig. 4; Supplementary table 1).
PERK интегрирует связанные с ER стрессы, чтобы обеспечить ослабление трансляции во время реакции на нецпакованные белки (UPR)108. UPR активируется как следствие накопления неправильно упаковенных или не упакованных белков в просвете ER, который является основным местом процессинга и укладки белка109. Активация PERK приводит к фосфорилированию eIF2α и к ослаблению трансляции, приводя в результате к снижению белковой нагрузки на ER. Нарушения регуляции функции PERK приводят к неспособности копирования при накоплении неправильно упакованных белков в ER, особенно в клетках с высокими секреторными запросами, таких как β клетки, приводя в результате к клеточным повреждениям и дисфункции110. Мутации гена, кодирующего PERK (EIF2AK3), вызывают редкое аутосомно рецессивное заболевание, Wolcott-Rallison syndrome (WRS)111. В соответствие с жизненно важной ролью PERK в секреторных клетках, наиболее выдающимся признаком WRS является диабет новорожденных или детей112. WRS также проявляет скелетные аномалии (epiphyseal dysplasia, osteopenia и spine defects) которые приводят к задержке роста, эпизодам острой печеночной недостаточности и умственной отсталостью, сопровождаемой микроцефалией и эпилепсией. Нокаут во всем теле Eif2ak3 у мышей воспроизводит фенотип WRS, включая диабет новорожденных, скелетные аномалии и задержку роста113,114. Специфическая делеция Eif2ak3 в инсулин-секретирующих β клетках вызывает дефекты пролиферации β клеток и дифференцировки во время плодного и неонатального периодов115,116. Неданвнее исследование показало, что вредные дефекты от делеции Eif2ak3 в поджелудочной железе вызываются повышенной экспрессией и передачей сигналов type I interferon receptor 1 (IFNAR1). Предполагается, что подавление IFNAR1 может способствовать смягчению клеточных повреждений, вызываемых дефицитом PERK117.
Диабет или фенотипические отклонения в нервной системе, частично перекрываемые с WRS, взываются мутациями в некоторы и др. генах, кодирующих ISR белки, напр., DnaJ homologue subfamily C member 3 (DNAJC3), immediate early response 3-interacting protein 1 (IER3IP1) и nucleotide exchange factor SIL1 (refs118-120) (Fig. 4). DNAJC3 является ко-шапероном теплового шока, который индуцируется с помощью ER стресса121 и который подавляет PERK, PKR122 и GCN2 (ref.123), чтобы супрессировать фосфорилирование eIF2α у удлинить трансляции в стрессовых условиях. Гомозиготные мутации в DNAJC3 вызывают усиление фосфорилирования eIF2α, приводящее к сахарному диабету (возраст начала 11-18 лет), к фенотипическими отклонениям в ЦНС (рано начинающаяся атаксия и признаки нарушения пирамидального тракта) и ПНС (сенсомотораня периферическая нейропатия и перцептивная тугоухость)119. Диабетический фенотип, воспроизводимый у Dnajc3 нокаутных мышей, обнаруживающих гипергликемию и глюкозурию, ассоциирует с апоптозом панкреатических β клеток и снижением уровней инсулина124. SIL1 является фактором обмена нуклеотидов для binding-immunoglobulin protein (BiP; известен также как HSPA5), который является критическим регулятором UPR. Мутации в SIL1 были обнаружены у пациентов с синдромом Marinesco-Sjogren, который характеризуется мозжечковой атаксией, задержкой развития, миопатией и катарактами118. IER3IP1 кодирует малый белок (~10 kDa), который располагается на ER и у частвует в реакции на ER стрессы. Он экспрессируется на высокм уровне в коре головного мозга и панкреатических β клетках. Мутации в IER3IP1 вызывают синдром микроцефалии с упрощенной gyration, эпилепсией и постоянным диабетом новорожденных (MEDS), который имеет перекрывающийся фенотип с WRS120.
Мутации в PPP1R15B нарушают активность фосфатазы PP1, приводя к повышению фосфорилирования eIF2α125,126. Клинически это вызывает диабет с ранним началом (в возрасте 15-28 лет), микроцефалию, умственную отсталость и дисплазию костей. Сходство клинического проявления среди родственных ISR нарушений подтверждает мнение, что любое отклонение от оптимального уровня фосфорилирования eIF2α или увеличивает (напр., из-за мутаций DNAJC3 и PPP1R15B) или снижает (посредством мутаций EIF2AK3), предпочтительно затрагивая β клетки, головной мозг и костные ткани. Эта гипотеза согласуется с фенотипами, вызываемыми мутациями в γ-суьъединице тримерного eIF2 комплекса127. Миссенс мутации в EIF2S3 (кодирующем eIF2γ) нарушают комплекс eIF2, вызывая mental retardation, X-linked, syndromic, Borck type (MRXSBRK (выраженный 'Marx Borck')), который характеризуется умственной отсталостью, микроцефалией, эпилепсией и задержкой роста127-129.
Одной из наиболее распространенных наследственных детских leukoencephalopathies является исчезновение белого вещества (VWM), которая также наз. детской атаксией с центральной гипомиелинацией130. VWM вызывается аутосомно рецессивными мутациями в любом из 5 генов, кодирующих eIF2B субъединицы (eIF2B1-eIF2B5) и характеризуется избирательным нарушением функции глиальных клеток головного мозга (олигодендроцитов и астроцитов), диффузно лишенных миелина и приводящих к кистозной дегенерации белого вещества131. Начинающаяся в раннем детстве прогрессирующая мозжечковая атаксия является характерным признаком VWM, но болезгь может возникать и в более зрелом возрасте и характеризоваться спастичностью, аторофией зрительного нерва с потерей зрения и эпилепсией. Начало болезни и быстрые нейрологические повреждения возникают вследствие стрессов, таких как небольшая травма головы и вызывающие лихорадку инфекции. Некоторые пациентки обнаруживают преждевременную неспособность яичников в дополнение к церебраьным аномалиям (ovarioleukodystrophy). Совместное появление неспособности яичников и нейродегенеративные нарушения описано также у индивидов с мутациями возможно в histidine-tRNA ligase, mitochondrial (HARS2) (ref.132) и возможно в leucine-tRNA ligase, mitochondrial (LARS2) (ref.133) при синдроме Perrault; и у индивидов с мутациями в alanine-tRNA ligase, mitochondrial (AARS2) при LKENP (leukoencephalopathy, progressive, with ovarian failure)134, указывая тем самым на общий механизм, лежащий в основе этих нарушений. Изучение трансгенных мышей, обладающих мутациями в eIF2B, подтвердили данные, полученные у людей, демонстрируя, что дисфункция астроцитов является главным своством VWM, приводящим к подавлению созревания олигодендроцитов и продукции миелина135,136. Механизм, вызывающий избирательный эффект мутаций eIF2B в глие, остается неизвестным, хотя недавнее исследование продемонстрировало повышенную чувствительность мышиных астроцитов к индуцированным мутациями в eIF2B нарушениям в митохондриальном оксидативном дыхании137. Повышение концентрации митохондрий достаточно, чтобы удовлетворить потребности в энергии в Eif2b5R132H/R132H эмбриональных фибробластах мышей (которые не участвовали в патогенезе VWM), но оказалось неспособным к этому в мутантных первичных астроцитах138.
Мутации в eIF2αкиназе GCN2 (кодируемой EIF2AK4), оказались сцепленными с pulmonary veno-occlusive disease (PVOD) и pulmonary capillary haemangiomatosis 21. PVOD является редкой формой легочной гипертензии, которая характеризуется гистологически фиброзной пролиферацией интимы septal вен и pre-septal венул, что приводит к сужению просвета и растяжению легочных капилляров и пролиферации 139. Обструктивные изменения в легочных венах вызывают увеличение сопротивления легочных сосудов, что у огромной фракции пациентов приводит к неспособности правого желудочка и к смерти (72% гибель в течение 1 года после диагноза) 140,141. Несмотря на доказательства аутосомно рецессивного наследования мутаций EIF2AK4 при PVOD и доступности мышиной модели, функциональная связь между GCN2 и патофизиологией болезни остается неизвестной. Некоторые доказательства подтверждают, что внешнесредовые стрессы играют критическую роль в запуске болезни 140. Остатется также неясным, почему мутации в GCN2 приводят к пульмональному фенотипу, тогда как др. ткани, обычно ассоциированные с нарушениями регуляции ISR, такие как головной мозг и поджелудочная железа, остаются незатронутыми. Недавнее исследование идентифицировало мутации в general control of amino-acid synthesis 1-like protein 1 (GCN1), активаторе GCN2, у индивидов с умственной отсталостью 142.
The mTOR pathway in human diseases
mTOR является serine/threonine protein киназой из семейства PI3K-related киназ, которые участвуют в двух белковых комплексах, состоящих измногих субъединиц, mTORC1 и mTORC2. mTORC1 интегрирует различные внутренние и внешние стимулы, чтобы скоординировать главные анаболические (напр., белковый синтез, липогенез и продукцию нуклеиновых кислот) и катаболические (напр., аутофагию) процессы в клетке. Открытие, что rapamycin (аллостерический ингибитор mTORC1) подавляет инициацию трансляции у разных видов, показало важность синтеза этого белка как главной нижестоящей мишени mTORC1 (refs143,144). eIF4E-binding proteins (4E-BPs), рибосомальные протеин S6 kinases (S6Ks), eIF4G, LARP1 и репрессора транскрипции Pol III MAF1 homologue (MAF1) действуют как непосредственные эффекторы mTORC1 в контроле белкового синтеза (Fig. 5). mTORC1 контролирует глобальный белковый синтез косвенно путем регуляции транскрипции и трансляции рибосомальных белков и факторов трансляции. Исследования животных, моделирующих рак, продемонстрировали, что нарушения белкового синтеза посредством фармакологических или генетических манипуляций существенно ослабляют онкогенный эффект активации mTORC1 145,146. Здесб мы рассмотрим болезни, связанные с двумя главными сигнальными путями, стоящими выше mTORC1, PI3K и пути узнавания аминокислот. Болезни , ассоцированные с на рушением регуляции др. регуляторов mTORC1 обсуждены в др. обзорах1 47,148.
The PI3K-AKT-mTORC1 pathway
Путь PI3K-AKT является главным сигнальным путем, стимулируемым факторами роста, который активирует mTORC1 аутем подавления tuberous sclerosis complex (TSC) (Fig. 5). Он также наиболее часто активирует сигнальные пути при раке; поэтому большинство рассматриваемых нарушений фактически связаны со склонностью к раку или сопровождаются доброкачественными или злокачественными опухолями. Нарушения регуляции пути PI3K-AKT-mTORC1 участвуют в большом количестве беолезней человека, включая метаболические заболевания, нарушения нейрального развития, синдромы извбыточного роста и иммунодефициты 149,150 (Fig. 5; Supplementary table 1). Некоторые широко распространенные онкогенные мутации пути PI3K-AKT-mTORC1 были также идентифицированы при нарушениях, не связанных с раковыми опуходями, указывая на возможность разработки многоцелевых лекарств для лечения рака и не раковых заболеваний 151,152. mTORC1 является главным регулятором клеточного роста. Соответственно, мвтации, которые активируют этот путь, сопровождаются разнообрзными фенотипами избыточного роста (Fig. 5; Supplementary table 1). Нарушения головного мозга часто обнаруживаются в результате нарушения регуляции пути mTORC1, подчеркивая важность этого пути для развития и функционирования головного мозга 153. Megalencephaly ассоциирует с мутациями PIK3CA, PIK3R2, PTEN, AKT3, TSC1, TSC2, MTOR и DEPDC5 (которые кодируют компоненты комплекса GAP activity towards Rag 1 (GATOR1) complex; see below) 154-156. Autism spectrum disorder (ASD), фокальная кортикальная дисплазия и судороги также часто наблюдаются при синдромах, связанных с mTOR 155,157,158.
PI3K
Мутации как в каталитической (p110) , так и регуляторной (p85) субъединицах класса IA PI3Ks обычно сцеплены с нарушениями у людей 149. Исход болезни обчыно зависит от тканевого распределения, а также от функциональных последствий мутаций (активирующих или супресирующих). В то время как мутации в повсеместно экспрессирующихся генах PIK3CA (кодирующего p110α) и PIK3R2 (кодирующего p85β) часто вызывают нарушения развития головного мозга 156, активирующие мутации в PIK3CD (кодирующем p110δ, иммуно специфическая изоформа p110) или её регуляторной субъединицы PIK3R1 (кодирующей p85α), вызывают синдром иммунодефицита, известный как активированный PI3Kδ синдром (OMIM: Immunodeficiency 14 and Immunodeficiency 36, соотв.) 159,160 (Fig. 5). Важно, что мутации потери функции PIK3CD или PIK3R1 оказались сцеплены с иммунодефицитом, указывая, что точная регуляция активности PI3Kδ необходима для соотв. функции иммунных клеток 2. Некоторые фенотипы иммунодефицита при мутациях избыточной функции устраняются воздействиями rapamycin 161, указывая, что активация mTOR играет критическую роль в патогенезе иммунодефицитаy.
PTEN: dose-dependent effects on disease
Опухолевый супрессор PTEN является lipid phosphatase, которая противодействует активности PI3K, приводя к подавлению фосфорилирования AKT (Fig. 5).Помимо своей основной роли при раке, мутации PTEN в зародышевой линии ассоциируют с несколькими самостоятельными синдромами избыточного роста и с ASD. Фенотипы, наблюдаемые у пациентов, несущих мутации PTEN соответствуют широкому диапазону тканей и тяжести, таким как макроцефалия, умственная отсталость, ASD, судороги, иммунодефицит и скелетные аномалии 155,162-167. Некоторые исследования предоставили неотразимые доказательства, что степень дефицита PTEN предопределяет тяжесть болезни. Частичная инактивация PTEN мутациями, которые делают белок нестабильным или снижают его способность супрессировать AKT, в основном ассоциированы с ASD и макроцефалией, тогда как полная инактивация PTEN ассоциирована с синдромами избыточного роста и раком 168.
AKT: the intriguing case of the E17K mutation
AKT имеет три изоформы, AKT1, AKT2 и AKT3, которые кодируются тремя генами. bВ то время как AKT1 экспрессируется повсеместно, AKT2 экспрессируется на наивысшем уровне в тканях, чувствительных к инсулину, а AKT3 экспрессируется на наивысшем уровне в головном мозге. Соматические активирующие мутации AKT1, E17K, вызывают синдром избыточного роста, известный как Proteus синдром 169. Интересно, что одна и та же мутациия E17K в AKT2 или AKT3 вызывает разные фенотипы. Мутация в AKT2 продуцирует метаболическое нарушение, известное как hypoinsulinaemic hypoglycaemia with hemihypertrophy (HIHGHH) 170, тогда как в AKT3, она ассоциирует с синдромом мегалэнцефалии (в частности, с megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome 2 (MPPH2)) 171. Перекрывающиеся и уникальные последствия гомологичных мутаций в AKT1, AKT2 и AKT3 подчеркивают важность тканевого распределения затронутого белка для проявления болезни. Поразительно, E17K в AKT1 также часто обнаруживается при раке. Время действия мутации (зародышевая линия в протививес саматической) и факторы предрасположенности (генетические или средовыые) предопределяют, будет ли E17K способствовать возникновению рака или незлокачественного фенотип. Также потеря AKT2 или AKT3 генерирует фенотипы, противополжные тем, что вызываются мутаций E17K в AKT2 или AKT3 (refs172,173).
Tuberous sclerosis complex
Tuberous sclerosis - это кожно-неврологическое заболевание, результат инактивирующих мутаций hamartin (TSC1) или tuberin (TSC2) (refs174,175); оно характеризуется доброкачественными опухолями во многих органах, судорогами, умственной отсталостью и ASD 176. TSC состоит из TSC1, TSC2 and TBC1 domain family member 7 (TBC1D7) и действует как GTPase активирующий белок для GTP-связывающего белка RHEB, чтобы подавлять активность mTORC1 (Fig. 5). Так, мутация TBC1D7 была идентифицирована у пациентов с макроцефалией, умственной отсталостью, дефектами костей и суставов, миопией и болезнями брюшной полости 177.
mTOR
Хотя мутации разных вышестоящих регуляторов mTORC1 оказались ассоциированы с разными болезнями, не связанными с раком, отсутствуют мутации компонентов mTORC1 (mTOR, regulatory-associated protein of mTOR (RAPTOR), proline-rich AKT1 substrate 1 (AKT1S1), target of rapamycin complex subunit LST8 и DEP domain-containing mTOR-interacting protein (DEPTOR)) не были идентифицированы до недавнего времени. Эта задержка была обусловлена редкостью таких мутаций, благодаря роли mTORC1 в развитии. Однако, недавнее секвенирование всего генома открыло de novo и соматические MTOR мутации у пациентов с магалэнцефалией, фокальной кортикальной дисплазией и эпилепсией 154,178.
Amino acid-dependent mTORC1 activation
В 2007 был описан новый синдром первичного иммунодефицита - иммунодефицит из-а мутации в MAPBP (mitogen-activated protein-binding protein)-interacting protein (MAPBPIP; известен также как LAMTOR2 и p14) - характеризующийся врожденной нейтропенией, дефицитом B и T клеток, гипопигментацией кожи и низким ростом 179(Fig. 5). Дефекты роста отличают синдром от лизосомных болезней, которые также характеризуются иммунодефицитом и гипопигментацией. MAPBPIP (p14) является компонентом пентамерного комплекса, наз. Ragulator 180. После активации с помощью аминокислот Ragulator рекрутирует RAS-related GTP-binding protein (RAG) GTPases на лизосомы, чтобы обеспечить активацию mTORC1 180-182. В дополнение к Ragulator, mTORC1 содержит несколько др. белковых комплексов, чтобы ощущать уровни лизосомных и цитозольных аминокислот, включая комплексы lysosomal vacuolar H+-ATPase (v-ATPase), GATOR1, GATOR2 и KICSTOR (kaptin (KPTN)-integrin-α FG-GAP repeat-containing protein 2 (ITFG2)-C12ORF66-seizure threshold 2 protein homologue (SZT2)) 183 (Fig. 5). Мутации в разных компонентах этих комплексов (за исключением GATOR2) оказываются сцепленными с разными нарушениями у людей. В частности, дефекты в GATOR1 или KICSTOR, которые являются ингибиторами mTORC1 в ответ на ограничение пищи, генерируют нейрологические фенотипы, сходные с наблюдаемыми при сцепленных с PI3K-AKT-mTOR нарушениях. Мутации, затрагивающие DEP domain-containing protein 5 (DEPDC5) (ref.184), NPRL2 и NPRL3 (ref.185) (компоненты комплекса GATOR1) и SZT2 (ref.186), KPTN 187 и C12ORF66 (ref.188) (компоненты комплекса KICSTOR) были идентифицированы у пациентов с судорогами, ASD, умственной отсталостью и нарушениями нейрального развития. v-ATPase является состоящим из многих субъединиц насосом протонов, который взаимодействует с комплексом Ragulator-RAG. Мутации в разных субъединицах v-ATPase оказались сцепленными с разными нарушениями у людей (Fig. 5; Supplementary table 1).
Other factors in neuronal diseases
Мутации регуляторных факторов трансляции, таких как GRB10-interacting GYF protein 1 (GIGYF1), GIGYF2, zinc-finger protein 598 (ZNF598) (компоненты комплекса репрессора трансляции 189) и eIF4G1 оказались сцепленными с различными нейродегенеративными и neurodevelopmental нарушениями, включая болезнь Parkinson 190-192, autism и умственную отсталость (Supplementary table 1). Кроме того, недавнее исследование подчеркнуло важность нарушений регуляции трансляции в ассоциированных с повторами нарушениях, таких как Fragile X syndrome и amyotrophic lateral sclerosis (ALS), это еще сильнее подчеркивает критическую роль контроля трансляции при нейродегенеративных болезнях 193-195.
The basis of phenotypic variability
Как действует нарушение регуляции общего синтеза белка, вызывающего такие разнообразные нарушения у людей? Несомненно некоторые факторы вносят вклад в это разнообразие. Во-первых,не все мРНК одинаково чувствительны к нарушениям регуляции трансляции. Различия последовательностей, длины и вторичной структуры 5'UTRs и 3'UTRs делают мРНК по-разному чувствительными к активности разных факторов трансляции. Напр., мРНК c длинными и структуированными 5'UTRs, связанные с митохондриями мРНК с короткими 5'UTRs и мРНК с 5'-терминальными олигопиримидиновыми мотивами чувствительны к нарушениям регуляции пути mTOR196-198. Дифференциальные реакции mTORC1-чувствительных субнаборов мРНК к разным факторам инициации трансляции, контролируют экспрессию функционально родственных транскриптов196. Кроме того, как было описано для mt-tRNA мутаций, различия в кодонах, используемых разными мРНК (напр., высокое содержание UUG при MT-ND6) может вносить вклад в исходы болезней (напр., при MELAS синдроме)36. Несмотря на наши знания нарушений регуляции трансляции немногих связанных с болезнью мРНК (GATA1 и MT-ND6), важный вопрос остаетс нерешенным. Во-первых, при каждой ли болезни имеются специфические мРНК , которые дерегулируются во время трансляции и ответственны за начало болезни? Во-вторых, какие свойства мРНК делают их более или менне чувствительнвми к нарушениям регуляции трансляции?
Кроме того, экспрессия генов варьирует и во времени и между тканями. Феноттипические отклонения при мутациях в генах, кодирующих AKT и PI3K, четко показывают, что тканевое распределение мутантных генов играет критическую роль в изсожах болезней, так как гомологичные мутации в трех изоформах AKT isoforms с 80% идентичных последовательностей ассоциированы с разными патлогиями (Fig. 6a). Потеря важных генов несовместима с эмбриогенезом. Т.о., мутации, вызывающие болезни, имеют тенденцию лишь частично снизить активность важных генов скорее, чем вызывать полную инактивацию или оказываются соматическими мозаичными мутациями. Различия в степени инактивации или распределения мутаций в теле вызывают разные патологические проявления, как напр., при PTEN и PI3Kδ (PIK3CD или PIK3R1) мутациях (Fig. 6b). Кроме того, разные ткани и типы клеток реагируют по-разному на нарушения регуляции трансляции. Ткани с высокимм потреблением энергии более склонны к нарушениям регуляции трансляции в митохондриях и периферические нейроны с длинными отростками более чувствительны к мутациям в ARS. Наконец, описанные для GCN2 мутации, вызывают предрасположенность к генетическим и средовым факторам, вносящими вклад в патогенез болезней.
 Fig. 6: Proposed mechanisms for tissue specificity of diseases caused by deregulation of protein synthesis.
Fig. 6: Proposed mechanisms for tissue specificity of diseases caused by deregulation of protein synthesis.
a | Tissue-specific gene expression has a crucial role in phenotype tissue specificity. This is exemplified by E17K mutations in the AKT genes. The figure was generated using the ProteomicsDB server203. b | The dose-dependent effect of PTEN inactivation is well established in cancer and may also explain the tissue-specific phenotypes observed with various PTEN mutations. PTEN partial inactivation often correlates with autism spectrum disorder (ASD) and macrocephaly, whereas complete inactivation correlates with overgrowth syndromes or cancer. Both loss-of-function and gain-of-function mutations affecting PI3K? activity can cause an immunodeficiency phenotype, suggesting that precise regulation of PI3K? activity is required for immune cell functions. There are multiple copies of mitochondrial DNA (mtDNA) in each cell, and most often, mitochondria carrying mutated (green) or wild-type (orange) copies are both present in each cell (heteroplasmy). Disease severity correlates with the amount of mutated mtDNA. c | Several models have been proposed to explain the vulnerability of neurons to translation deregulation57. The prevailing hypothesis postulates that a subset of functionally related mRNAs is stored in distal compartments of neurons (dendrites and axons). These localized mRNAs engage in translation only if they receive the proper internal (arrowheads) and/or external (arrows) cues. Although such remote regulation of translation is beneficial for supporting abrupt neuronal response, the long distance between the nucleus and synapses renders neurons hypersensitive to translation deregulation. ER, endoplasmic reticulum; HIHGHH, hypoinsulinaemic hypoglycaemia with hemihypertrophy.
Нервная система особенно уязвима к нарушениям регуляции трансляции мРНК, скорее всего, благодаря структурным и метаболическим свойствам. Нейроны имеют чрезвычайно сложную и поляризованну морфологию, в которой дендриты и аксоны являются отличающимися функцимональными компартментами и эти компартменты зависят от локальной трансляции предсуществующих, локальных мРНК, быстро реагирующих на комплеас стимулов (Fig. 6c). Удаленная регуляции трансляции мРНК делает нейроны более чувствительными к нарушениям трансляции. Кроме того, глюкоза является основным источником энергии в головном мозге. Хотя головной мозг составляет лишь 2% от веса тела, он потребляет наивысшие количества глюкозой-производимой энергии (20%) по сравнению с др. органами199. Поэтому вполне возможно, что нарушения трансляции мРНК, чьи продуктыучаствуют в метаболизме глюкозы преимущественно и приводят к нарушениям в головном мозге. Это согласуется с наблюдением, что некоторые нарушения, связанные с развитием нервной системы, вызываются нарушениями регуляции трансляции и ассоциируют с дефектами в метаболизме глюкозы и с диабетом, как и в случае болезней, связанных с нарушениями регуляции ISR и mTOR пути. Разработка терапевтических подходов, нацеленных на метаболизм глюкозы (таких как ketogenic диета200 и противо-диабетические лекарства201) могут оказаться многообещающими стратегиями лечения нейрологических нарушений, вызываеых нарушениями регуляции трансляции.
Понимание патофизиологии редких болезней человека может дать преимущества при использовании индуцированных плюрипотентных стволовых клеток202 в комбинации с широко используемыми техниками редактирования генов (напр., CRISPR-Cas9), чтобы понять трансляцию на геномном уровне (напр., используя профилирование рибосом).
|